
Abstract
Hypermobility spectrum disorders (HSDs) encompass an array of connective tissue disorders characterized by joint instability and chronic pain. Fatigue and other systemic symptoms that affect daily functioning may occur, as well. Accurate data on incidence and prevalence of HSDs is hampered by lack of awareness of these conditions and the wide heterogeneity of their clinical presentation. Identifying which type of HSD is present is important in guiding appropriate care. In particular, making the diagnosis of hypermobile Ehlers-Danlos syndrome (hEDS) is important, as individuals with hEDS may be at risk for more significant multisystem involvement. Diagnostic criteria for hEDS include measures of joint hypermobility, skin and other connective tissue findings, and lack of evidence of a different type of Ehlers-Danlos syndrome. Beyond accurate diagnosis, HSDs pose many challenges for primary care providers, as ongoing patient education, patient empowerment, and coordination of a multidisciplinary treatment team are integral to proper care. This article describes the incidence and prevalence, pathophysiology, diagnosis, and management of HSDs, including clinical cases exemplifying how joint hypermobility might present within a primary care setting.
Case 1
Jennifer is a 15-year-old female who comes in with her mother to discuss her diffuse muscle and joint pain and “feeling tired all the time.” She states that the pain is most intense in her shoulders, back, feet, and ankles. The pain has been present since “at least middle school” and has increased since starting high school. She has a history of recurrent shoulder dislocations starting at age 13. She also describes 1 episode of a patellar dislocation with normal x-rays. She describes herself as “double jointed.” She has also sprained her ankles multiple times.
She gets 8 to 10 hours of restful sleep every night but still is barely able to make it through the school day without falling asleep, much less get home and do her homework. Her past medical history includes anxiety, chronic headaches, gastroesophageal reflux disease, irritable bowel syndrome (IBS), and orthostatic hypotension. She feels lightheaded when getting up quickly and has had an electrocardiogram that was normal. She has had an umbilical hernia repair as well as a revision. Her mother also has joint laxity, anxiety, and IBS.
Her vitals are normal. She can extend both elbows and knees to greater than 10 degrees, extend fifth metacarpophalangeal (MCP) joints past 90 degrees, and easily put the palms of her hands on the floor without bending her knees (Beighton score 7/9). The scar on her abdomen seems atrophic. Her skin is soft and mildly hyperextensible. There is livedo reticularis (skin translucency revealing subcutaneous veins). She demonstrates arachnodactyly, with thumbs and fifth fingers able to meet around the contralateral wrist (Walker wrist sign). There are pale stretch marks at the lateral hips, breasts, and axillae; she reports these developed during her adolescent growth spurt.
Case 2
Sonja is a 23-year-old woman who presents with widespread pain, headaches, insomnia, and anxiety, which began 18 months ago after a motor vehicle accident (MVA). Before the accident she had been in very good health, enjoyed long-distance running, worked at a coffee shop, and was enrolled in undergraduate studies. She did have some preaccident wrist, hand, and elbow pain associated with computer use. She suffered no major injuries in the MVA aside from whiplash, but months after the accident was still having significant neck pain and headaches as well as a range of new and disconcerting symptoms such as upper back pain, foot pain, tinnitus, chest pain, photophobia, and phonophobia.
Since that time, she had been referred to and evaluated by specialists in multiple disciplines including neurology, rheumatology, physical therapy, and sports medicine. She also had a variety of laboratory tests and imaging studies, all of which were normal. She had tried numerous medications including duloxetine, amitriptyline, naproxen, acetaminophen, and gabapentin. However, her symptoms worsened, and she began to experience significant fatigue. She started working with chiropractors, getting adjustments several times per week to “keep everything in place.” The adjustments gave partial relief, but only for a day or 2.
She left her job and took a leave of absence from school after the MVA and has been unable to return. She moved back in with her parents and spends much of the day sedentary, aside from short walks that leave her feeling achy and fatigued. She does not use alcohol, tobacco or other forms of nicotine, or illicit drugs.
She has no known family history of joint hypermobility.
On examination, she is somewhat anxious and expresses concern over her vertebrae being “out of place,” as communicated by her chiropractor. She has a body-mass index of 20 and resting tachycardia at 105 beats per minute; other vital signs are normal. She has difficulty tracking during testing of extraocular movements, citing eye fatigue, but has no nystagmus. She has pain with neck motion in all planes. She has normal spinal range of motion but experiences spine pain with thoracic and lumbar flexion and side bending. Effort during manual muscle testing is limited due to pain, but she is able to mount grade 5 (normal) effort on repeated testing. Her Beighton score is 4/9, with points for fifth finger MCP hyperextension and elbow hyperextension bilaterally. Her skin is of normal softness and elasticity. There is no arachnodactyly.
Joint Hypermobility and Hypermobility Spectrum Disorders—Definitions
Joint hypermobility is a common complaint seen in primary care. Joint hypermobility refers to increased active or passive movement of a joint beyond its normal range. One can have joint hypermobility without having a hypermobility spectrum disorder (HSD). Joint hypermobility can be categorized by question-based and physical examination-based measures. On a validated, commonly used patient questionnaire eliciting hypermobility symptoms, an affirmative answer to 2 or more out of 5 questions shows 84% sensitivity and 80% specificity for joint hypermobility (Table 1). The physical examination-based counterpart to this questionnaire is the Beighton hypermobility score (Table 2), which is commonly employed in the assessment of the hypermobile patient.3 Though the Beighton hypermobility scoring rubric was initially introduced as an instrument for epidemiologic research rather than a tool to be used diagnostically, it has now come to be used as a measure of generalized hypermobility.2 Higher scores on the Beighton scale simply represent a greater number of affected joints rather than a higher degree of laxity.
Validated Questionnaire for Joint Hypermobility
Beighton Hypermobility Score—Maximum Score = 9 Points
Hypermobility Spectrum Disorder—Diagnostic Criteria
Kirk and colleagues first described hypermobility syndrome in 1967, describing a syndrome of familial ligamentous laxity resulting in recurrent joint pain and periodic joint effusion.4 It was thought to be isolated to the musculoskeletal system, separate from Marfan syndrome and the Ehlers-Danlos syndromes (EDSs), and to occur in otherwise healthy people. Over time, overlap in signs and symptoms has been noted with other hereditary disorders of connective tissue. There was considerable overlap between hypermobility syndrome and hypermobility-type (type III) EDS (hEDS), to the point that differentiating the 2 entities was very difficult (Figure 1).
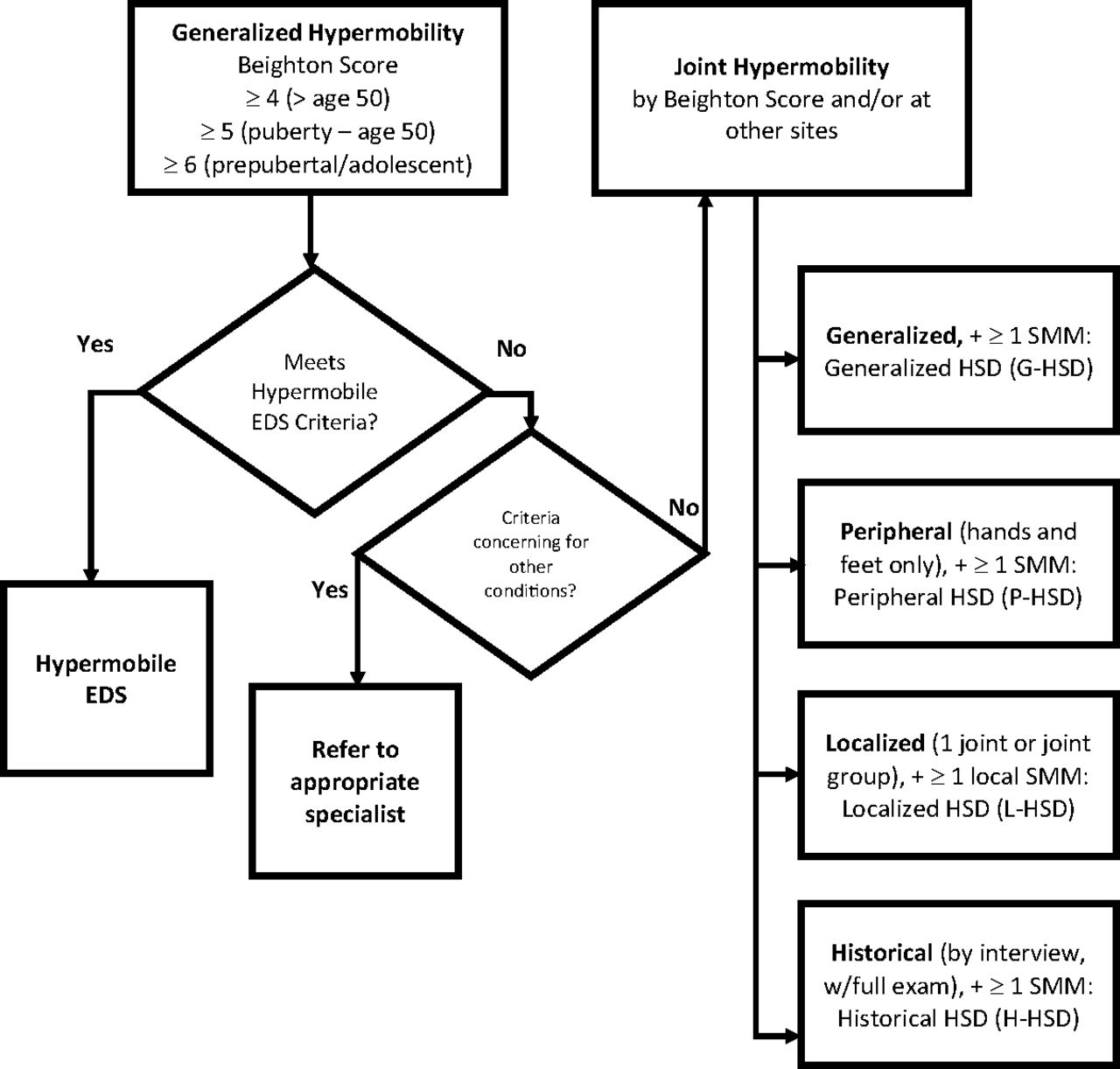
Diagnosing disorders of joint hypermobility: distinguishing hypermobile Ehlers-Danlos syndrome and the hypermobility spectrum disorders. Abbreviations: hEDS, hypermobile Ehlers-Danlos syndrome; EDS, Ehlers-Danlos syndrome; HSD, Hypermobility spectrum disorder.
From 2000 until 2017, patients with hypermobility were diagnosed with what was then called benign joint hypermobility syndrome (JHS), once the presence of another connective tissue disorder had been excluded. In 2017, JHS was reclassified and renamed the hypermobility spectrum disorders,5 which are subcategorized into generalized (G-HSD), peripheral (P-HSD), localized (L-HSD), and historic (H-HSD) types. At the same time, type III EDS was renamed hypermobile EDS (hEDS) and given a new set of diagnostic criteria that better differentiate it from G-HSD (Table 3). If significant hypermobility is present on the Beighton hypermobility score, but a patient does not fit the criteria for hEDS or another of the EDSs, the patient is diagnosed as G-HSD. Yet, the 2 disorders can remain difficult to clinically differentiate. For example, the confirmation of a positive family history (or lack thereof) may flip the diagnosis from 1 to the other.
Diagnostic Criteria for Hypermobile Ehlers-Danlos Syndrome (hEDS)3
The HSDs are defined by joint hypermobility using the Beighton score (Table 2) plus 1 or more secondary musculoskeletal manifestations including post-traumatic symptoms, pain, altered proprioception, and a variety of other characteristics like pes planus, kyphosis, scoliosis, or joint misalignment.6 The designation should be used for individuals with symptomatic joint hypermobility when criteria are met and when hEDS has been ruled out. H-HSD is diagnosed in cases of documented prior hypermobility that has disappeared with increased age.7
Hypermobility Spectrum Disorders—Incidence and Prevalence
Accurate characterization of the incidence and prevalence of HSDs and hEDS has been hampered by a lack of its recognition, in part due to the often-benign nature of the condition and the fact that it may improve rather than worsen with aging, as well as by a previously unclear distinction in the literature between HSD and hEDS. Prevalence of generalized joint hypermobility among college-aged students ranges from 12.5% to 26%.8,9 In these college-aged populations, the incidence of hypermobile joints was higher than that in older adults. A more recent UK symptom-based survey estimated prevalence of HSDs in a general population at 3%.10
Genetics in Hypermobility Spectrum Disorders
HSDs have a strong genetic basis but no known genetic markers. The vast majority of cases do not seem to be linked to any identifiable mutation, although pedigree studies have shown a weak autosomal dominant inheritance pattern with variable penetrance.10,11 The EDSs comprise a heterogeneous group of rare monogenic conditions caused by genetic defects in an array of extracellular matrix genes within soft connective tissues. The genetic basis of hEDS remains unknown, in contrast to other well-described EDS subtypes. In view of the considerable clinical overlap with HSDs, many consider HSDs and hEDS to be a single clinical entity, with variable manifestations possibly reflecting variability in penetrance.12
The Pathogenesis of Pain in HSDs
The specific underlying causes and mechanisms of pain in HSD remain poorly understood. Factors likely contributing to the generation and chronicity of pain include nociceptive pain due to structural changes in affected joints, muscle, and connective tissue; neuropathic pain; impaired proprioception and muscle weakness; and central sensitization. These mechanisms are not mutually exclusive and may all contribute to pathogenesis.12 Current theories emphasize the importance of localized biomechanical overloading and chronic soft tissue injury due to joint laxity and instability. Repetitive microtrauma may lead to altered kinematics, which, in turn, cause overload on other joints and further soft tissue injury manifesting as arthralgias and diffuse musculoskeletal pain.10,13 HSD patients tend to lack proprioceptive acuity, which may increase risk of injury.14 In addition, patients tend to have decreased muscle mass and muscle strength, potentially due to increased laxity of tendons, which are unable to transmit power produced by muscles.15,16 Altered biomechanics and posture can place overstress on muscles and fascia, causing additional pain.
In addition to these biomechanical abnormalities, patients with HSD may have neurologic disturbances that contribute to diffuse musculoskeletal pain. Generalized hyperalgesia (fibromyalgia) is a common complaint and is believed to be mediated by central sensitization. The biochemical underpinnings of hyperalgesia in HSD are unknown. Some have speculated that it serves an adaptive purpose as a compensatory mechanism to prevent joint instability.17 Fear of provoking pain and injury may lead to avoidance of activity, ultimately resulting in deconditioning and exercise intolerance. This often aggravates functional disability and leads to anxiety and depression symptoms, which in turn reduce pain tolerance and promote further pain.16,18
Skin Manifestations
Individuals with HSD and hEDS can display a variety of dermatologic manifestations related to abnormal connective tissue (Table 4). Bruising and bleeding in people with HSD occur due to fragile skin and capillaries rather than abnormal coagulation.19
Skin Manifestations of HSD and hEDS19
Joint Findings in Patients with HSDs
Patients present with generalized joint hypermobility with or without joint laxity. While these 2 terms are often used interchangeably, laxity indicates instability, whereas hypermobility denotes an increase in the joint's range of motion beyond normal.20 Weight-bearing joints are more common sources of pain. Examination may reveal articular or periarticular tenderness, but significant synovitis should prompt reconsideration of the diagnosis and investigation for inflammatory conditions. Small noninflammatory effusions may occur due to repetitive stress on joint structures, and joint deformity may occur and can mimic inflammatory arthritis. Children and young adults may present with scoliosis, pes planus, genu valgum, genu recurvatum, and/or patellar dislocation/subluxation.10
People with HSDs often present with a variety of multisystem complaints (Table 5).
HSD in Pregnant Women
Pregnant women present a unique subset of patients with HSDs in the context of the increased joint laxity normally seen in pregnancy. There is a paucity of research to guide clinicians, but no evidence of increased risks of adverse pregnancy outcomes.22,23 There is some elevated risk of late-pregnancy back or sacroiliac pain, along with some risk of early labor, in hEDS and probably in the HSDs. Dysautonomia symptoms often improve as maternal blood volume increases in later pregnancy. Given evidence of an autosomal dominant inheritance pattern with variable penetrance, children of HSD patients may have up to a 50% chance of hypermobility of some degree.
Affective Disorders (the Neurocognitive Phenotype)
A positive correlation exists between HSDs and anxiety. Joint hypermobility is more common in patients diagnosed with anxiety,24 and anxiety is disproportionately present in people with joint hypermobility.25,26
In a study of patients with joint hypermobility, anxiety was found in 70% of patients in comparison with a 20% incidence in the age- and gender-matched control group. In the follow-up reverse case-control study, joint hypermobility was found at 17 times the incidence in patients diagnosed with anxiety as compared with age- and gender-matched controls without an anxiety diagnosis.27 This correlation between affective state and heritable hypermobility conditions has recently been labeled the “neurocognitive phenotype.” Both neuroimaging and genetic studies point to the depth of the interconnectedness of structure and affect.
Eccles and colleagues found that participants with joint hypermobility had significant differences on magnetic resonance imaging (MRI) in brain regions associated with anxiety in comparison to nonhypermobile participants. Hypermobile subjects had significantly enlarged amygdalae bilaterally in comparison to the nonhypermobile cohort.28 In addition, the authors found structural differences in the anterior cingulate cortex, another brain structure associated with cognitive control of emotions and a driver of autonomic arousal.
Dysautonomia and Exercise Intolerance
Problems with autonomic function, such as orthostatic intolerance (ie, postural orthostatic tachycardia syndrome), body temperature dysregulation, and exercise intolerance, are common in people with HSD.29,30 While physical activity is often recommended for patients with anxiety, such activity in anxious people with HSD may not have the intended effect. It has been hypothesized that prior unpleasant experiences with physical activity contribute to the kinesiophobia reported in people with joint hypermobility29; therefore, like patients with fibromyalgia, people with HSD may not enjoy the anxiolytic benefits of exercise. Coupled with the postexercise malaise that many people with HSD report, a blanket recommendation to increase physical activity may actually exacerbate symptoms. In working with patients who report anxiety, screening for joint hypermobility may offer insights into effectively treating their symptoms of anxiety and in compassionately supporting their desire for well-being. Physical activity can be safely initiated even in anxious hypermobile patients if started at low intensity, titrated slowly in a supportive environment, and targeted to the particular biomechanical problems and exercise needs of the individual patient. Support and assistance from behavioral health professionals may be necessary as exercise is introduced.
Challenges in Primary Care
The primary care provider treating hypermobility syndromes faces many challenges. These may include sorting through multiple and often vague complaints, considering competing diagnoses, determining and often triaging an appropriate workup based on this differential, coordinating a multidisciplinary treatment team, and providing patient education and appropriate self-educational resources once a diagnosis is established.
The patient with HSD will often have a range of complaints involving multiple organ systems. Many of these are not included in the validated questionnaire for hypermobility, and these are often the symptoms that draw the attention of patients and cause them to seek care. These complaints may include manifestations of autonomic dysfunction and other cardiovascular concerns,29 a wide range of gastrointestinal issues,21 anxiety, phobic disorders, major depression,25 headaches and chronic fatigue,10 among others. When evaluating a patient with such a wide array of complaints, one may wish to focus on the individual concerns that seem more serious in nature to the provider and those that are most bothersome to the patient.
Differential Diagnosis
While the initial workup for HSD involves eliminating other potential etiologies, the diagnosis is ultimately clinical and requires no laboratory or imaging tests for confirmation. If a patient presents to the primary care clinic with complaints that suggest HSD, obtaining previous records can prove an important step to avoid a previously completed and costly workup. Using the aptly named “A Simple Questionnaire to Detect Hypermobility” can help streamline an initial evaluation, as it has a sensitivity of 84% and specificity of 85% for HSD.1 Family history can provide further important information, especially if a first-degree relative has an unequivocal diagnosis of HSD. Identification and/or exclusion of fibromyalgia, complex regional pain syndrome, nerve impingement syndromes, and non-HSD conditions causing hypermobility are important in the evaluation.2
HSD and hEDS exist on the same physiologic continuum, but hEDS manifests at the more symptomatic edge of the spectrum. Patients with hEDS generally present to a physician's office at a younger age, have more severe symptoms, and must conform to strict diagnostic criteria put forth in 2017 by the International Consortium on Ehlers-Danlos Syndromes and Related Disorders.3 However, once a diagnosis is made, there is no difference in the approach to treatment for HSD and hEDS (Table 6).
Comparing HSD, hEDS, and Other EDS Disorders
Treatment Options
Once the diagnosis of HSD or hEDS is suspected or confirmed, the majority of patients can be managed in the primary care setting with a multidisciplinary approach that involves physical therapy, biofeedback, cognitive behavioral therapy/psychotherapy, and pharmacotherapy.31 The tenets of pharmacotherapy are similar to those in most chronic pain conditions with avoidance of opioids, preference for nonsteroidal anti-inflammatory drugs (NSAIDs)/acetaminophen, topical NSAIDs, lidocaine patches where neuropathic pain is present, antidepressants (particularly SNRIs or low-dose tricyclic antidepressants [TCAs]), and/or anticonvulsants.31 Symptomatic treatment of muscle pain or spasm may be treated with antispasticity agents (ie, baclofen or tizanidine). Patients with comorbid fibromyalgia may benefit from gabapentin or pregabalin. Patients with exercise intolerance or orthostatic symptoms may not tolerate TCAs or tizanidine.
Successful treatment of chronic pain requires a multidisciplinary approach and should focus on treating the cause of the pain (eg, dislocation of a joint, muscle imbalance with spasm, sprain/strain), reducing pain where possible, and maximizing functional capacity and quality of life. It should also be noted that fatigue and pain seem to be linked, leading to decreased quality of life. Treating dysautonomia in patients with HSD may improve both fatigue and pain.32
Lifestyle modifications, regular exercise without overtraining, and stretching techniques focused on tight muscles (but not lax joints) may help. Physical and occupational therapy programs centered on strengthening the core and periarticular musculature can improve proprioception, reduce injury, and reduce pain while improving function (see Table 7).
Conclusion
Primary care clinicians will frequently see patients with hypermobility syndrome disorders. Recognition of the diagnostic criteria, common overlaps, and differentiating features of the various disorders under this umbrella will help clinicians appropriately diagnose patients and develop multidisciplinary treatment approaches to optimize outcomes.
Notes
This article was externally peer reviewed.
Funding: None.
Conflict of interest: None.
- Received for publication July 21, 2020.
- Revision received November 17, 2020.
- Accepted for publication November 17, 2020.
References
In this issue



{kind=link}
Jump to section
Related Articles
Cited By...
- One-year effectiveness of high-load compared with low-load strengthening exercise on self-reported function in patients with hypermobile shoulders: a secondary analysis from a randomised controlled trial
- The Most Frequently Read Articles of 2021
- Family Medicine Research on Health Equity, Addiction, and Eating Breakfast--Just for Starters