
Wegener granulomatosis (WG) is an uncommon condition characterized by necrotizing granulomatosis of the upper and lower respiratory tract and glomerulonephritis. We describe a case of a 42-year-old white man with symptoms of pansinusitis who gradually developed more symptoms of WG. This case emphasizes early diagnosis and treatment to avoid further complications.
Case Report
The patient was a 42-year-old white man with a history of sinusitis for almost 2 years. He was treated with different antibiotics as an outpatient without any improvement. He developed a lung mass on chest radiograph, so he was referred to the surgical service for biopsy. On the day of the planned biopsy, the patient was febrile with an elevated white blood cell count (WBC). He also had severe headache; therefore, biopsy was canceled and the patient was transferred to the Family Practice team.
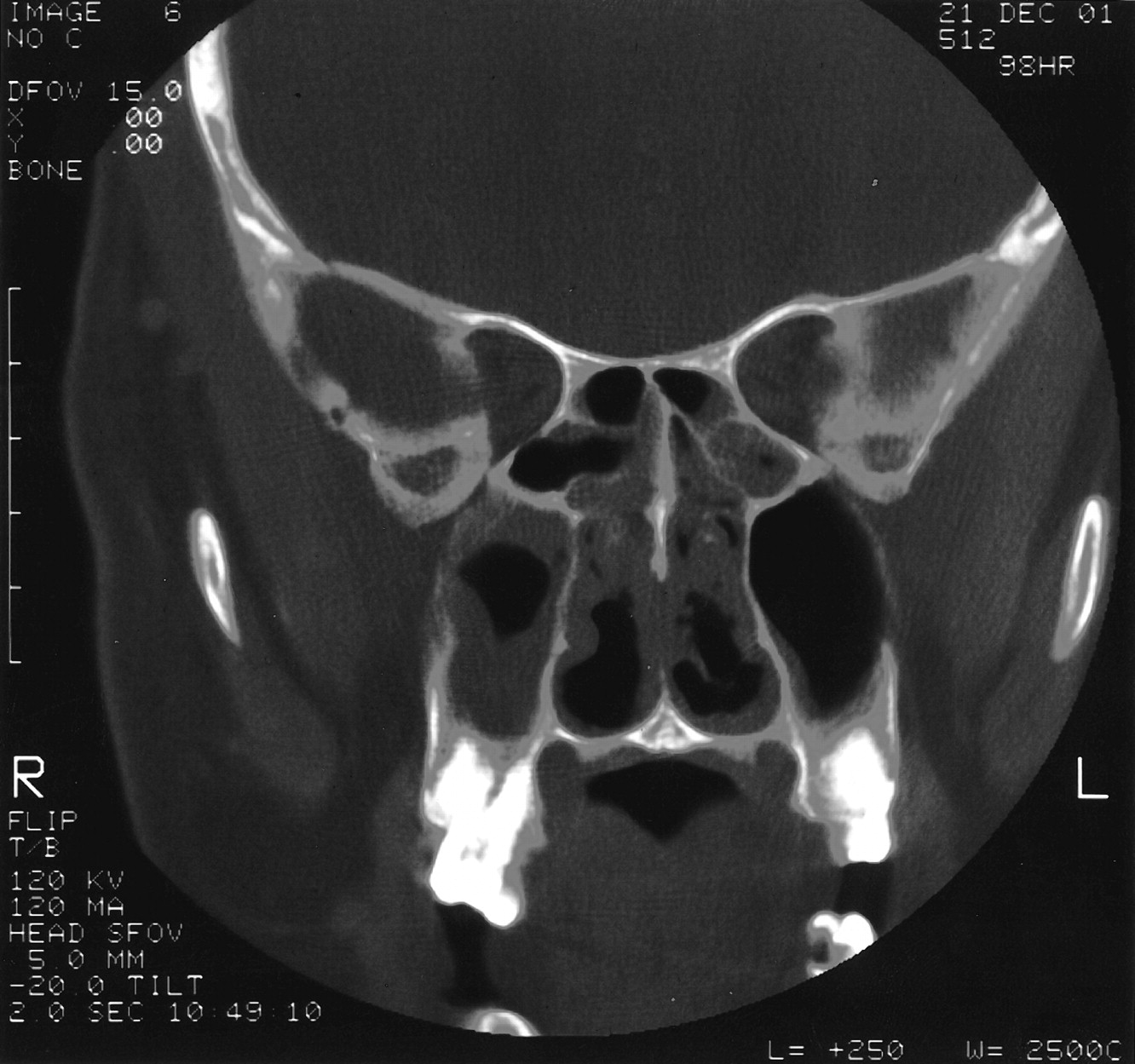
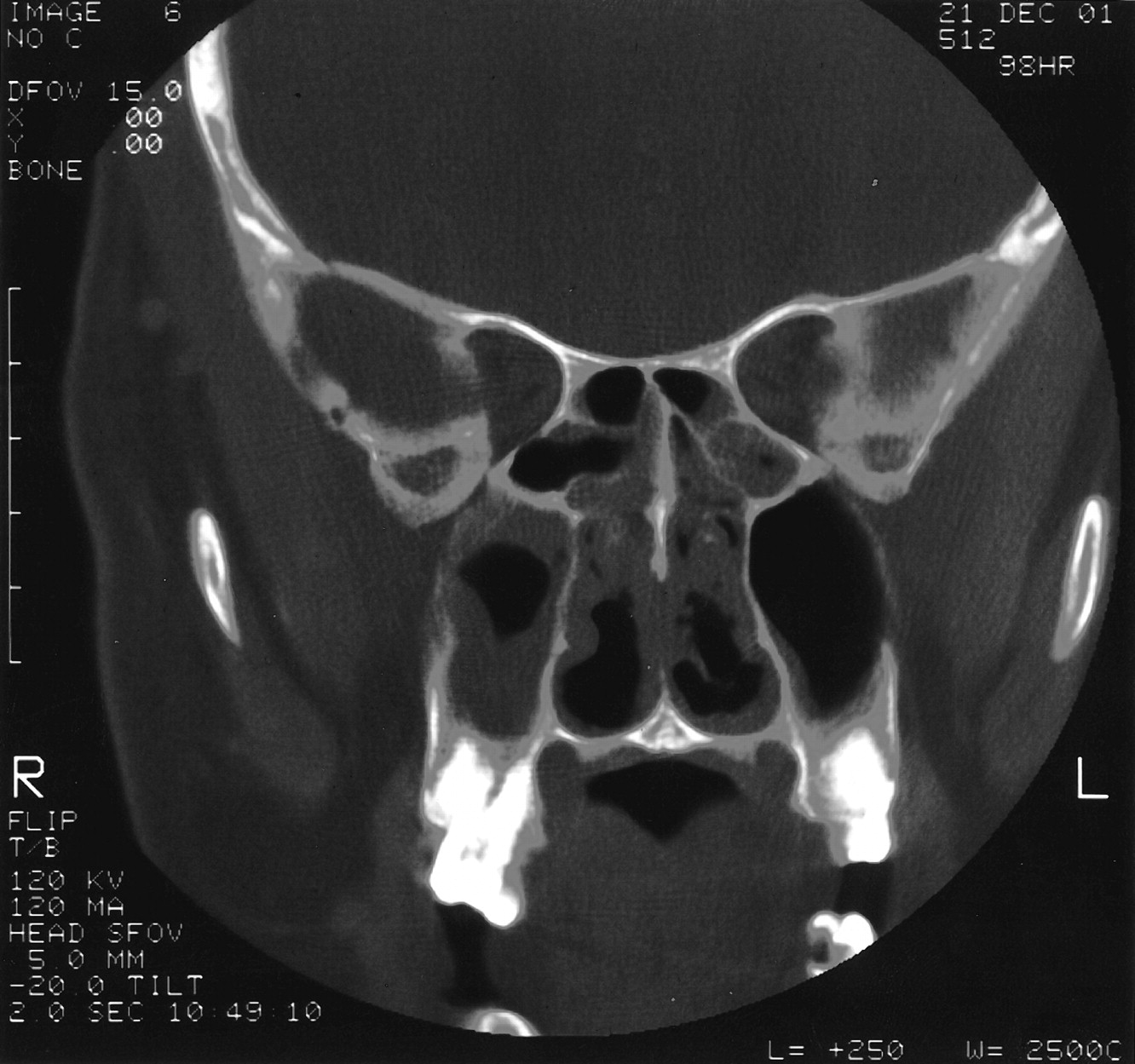
At admission, his temperature was 98.8°F, pulse was 90 beats/min, respiratory rate was 20 breaths/min, blood pressure was 150/90 mm Hg, and his weight was 217 pounds. He was not in severe distress. Tympanic membranes were clear with positive light reflexes. His throat was erythematous and without other lesions. His neck was supple with no meningeal signs. His lungs were clear to auscultation bilaterally. His abdomen was neither tender nor distended. There was no edema, cyanosis, or clubbing of his extremities. Computerized tomography (CT) of his sinuses showed mucosal thickening in all sinuses, including sphenoid, frontal, ethmoidal, and maxillary (Fig. 1). Laboratory studies included: WBC, 12.2 103/μL; hemoglobin, 12.3 g/dL; hematocrit, 0.345; platelets, 271 103/μL; serum urea nitrogen, 21 mg/dL; creatinine, 0.9 mg/dL; aspartate aminotransferase, 39 U/L; alanine aminotransferase, 129 U/L; alkaline phosphatase, 91 U/L.
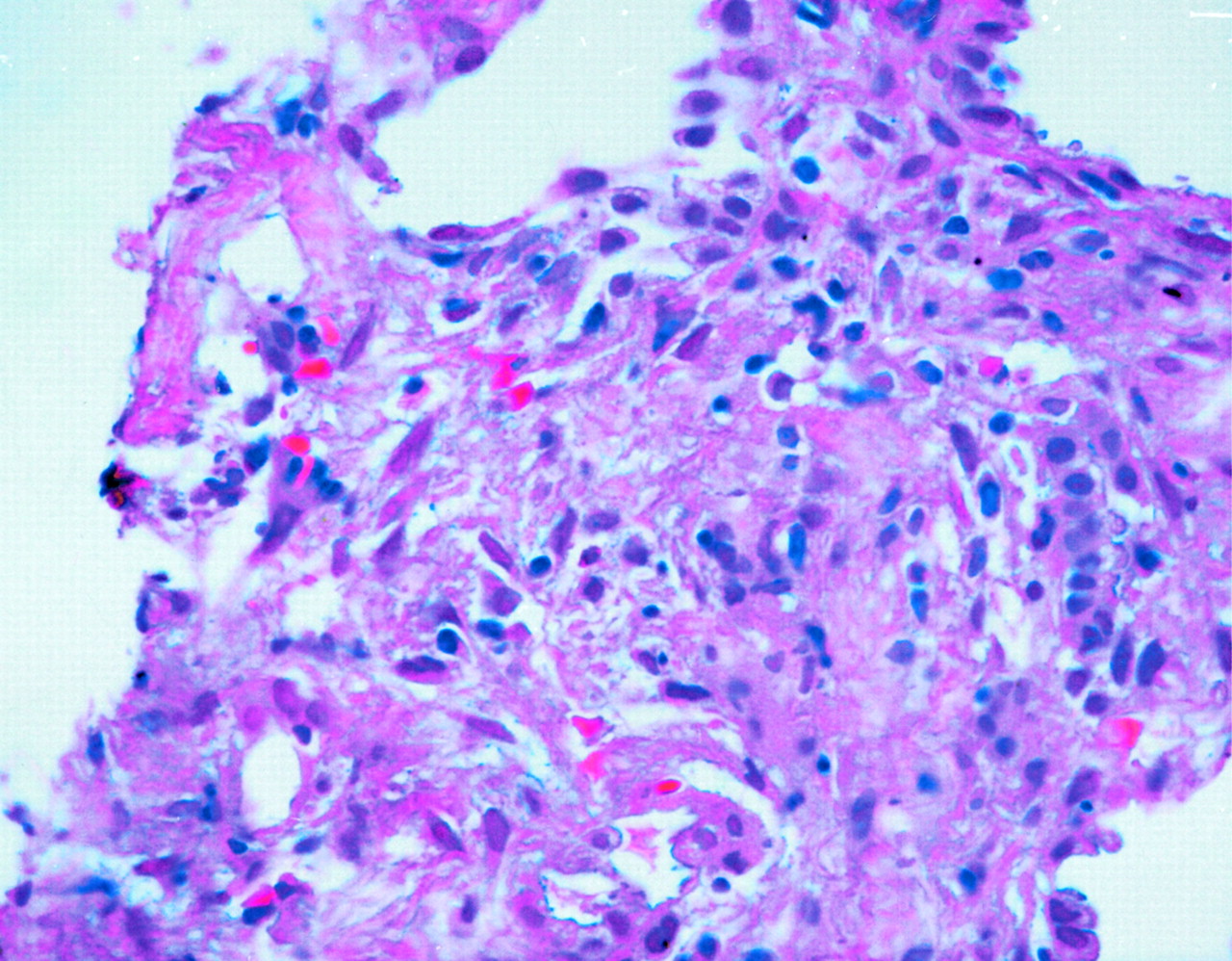
Focus of granulomatous inflammation, lung (hematoxylin and eosin; original magnification, 400×).
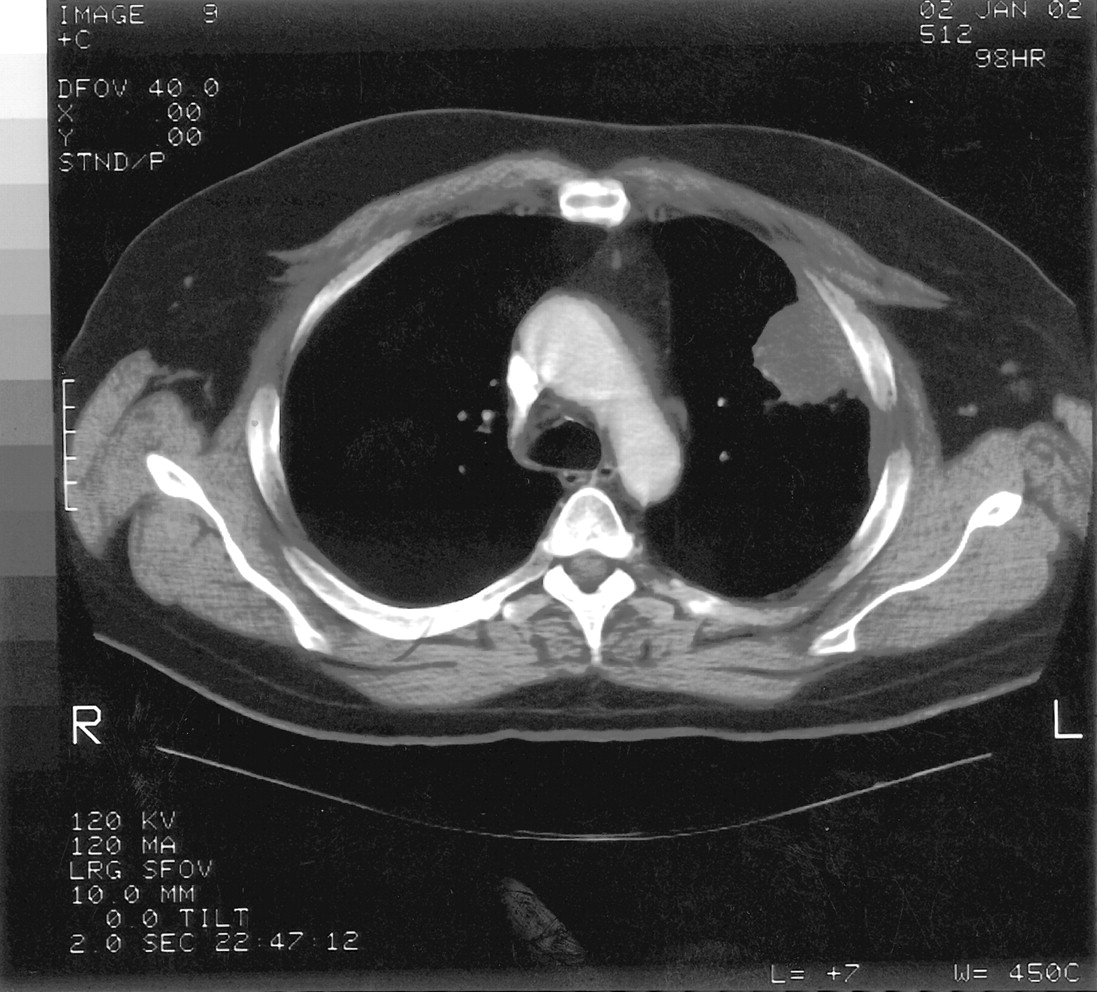
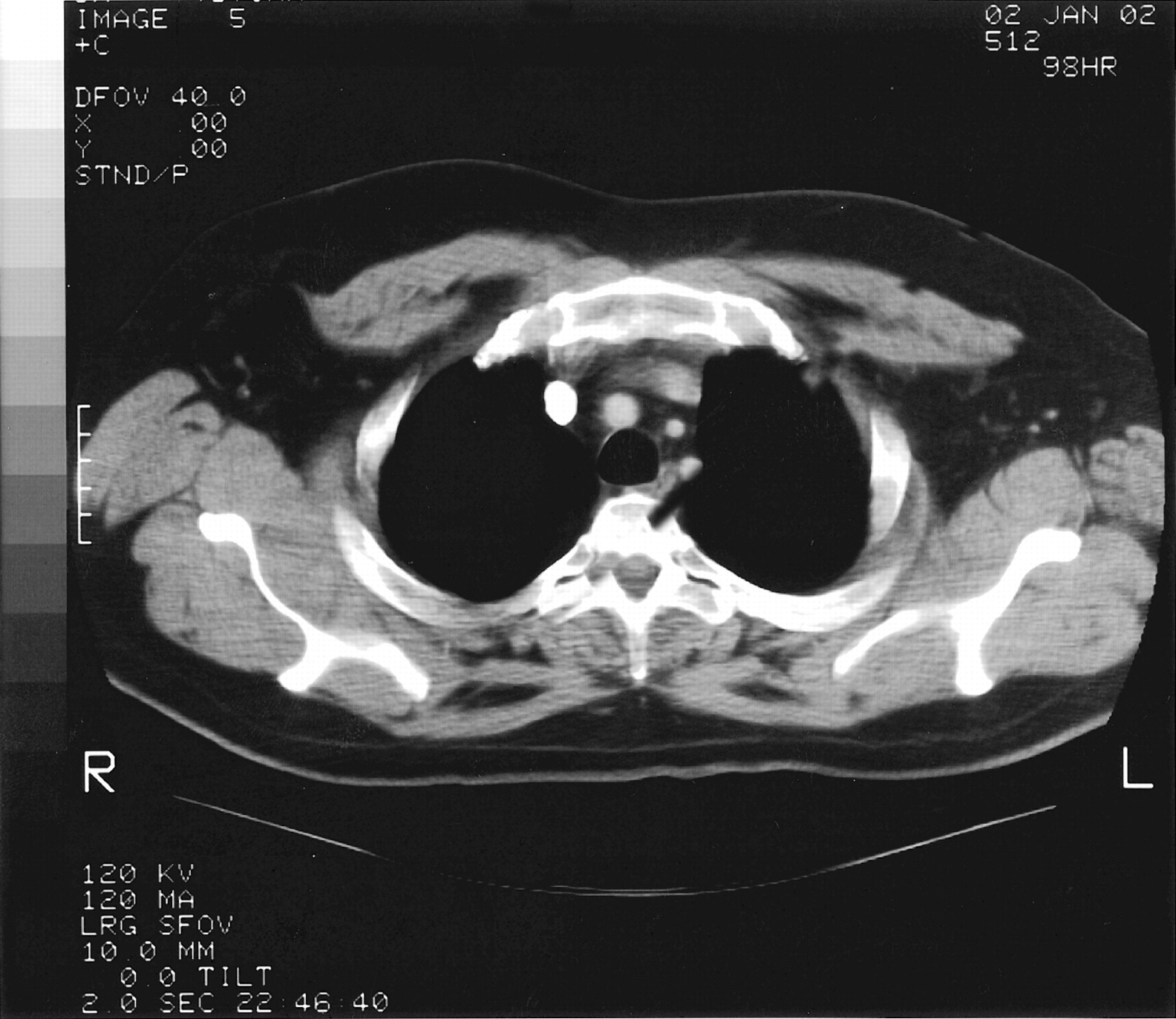
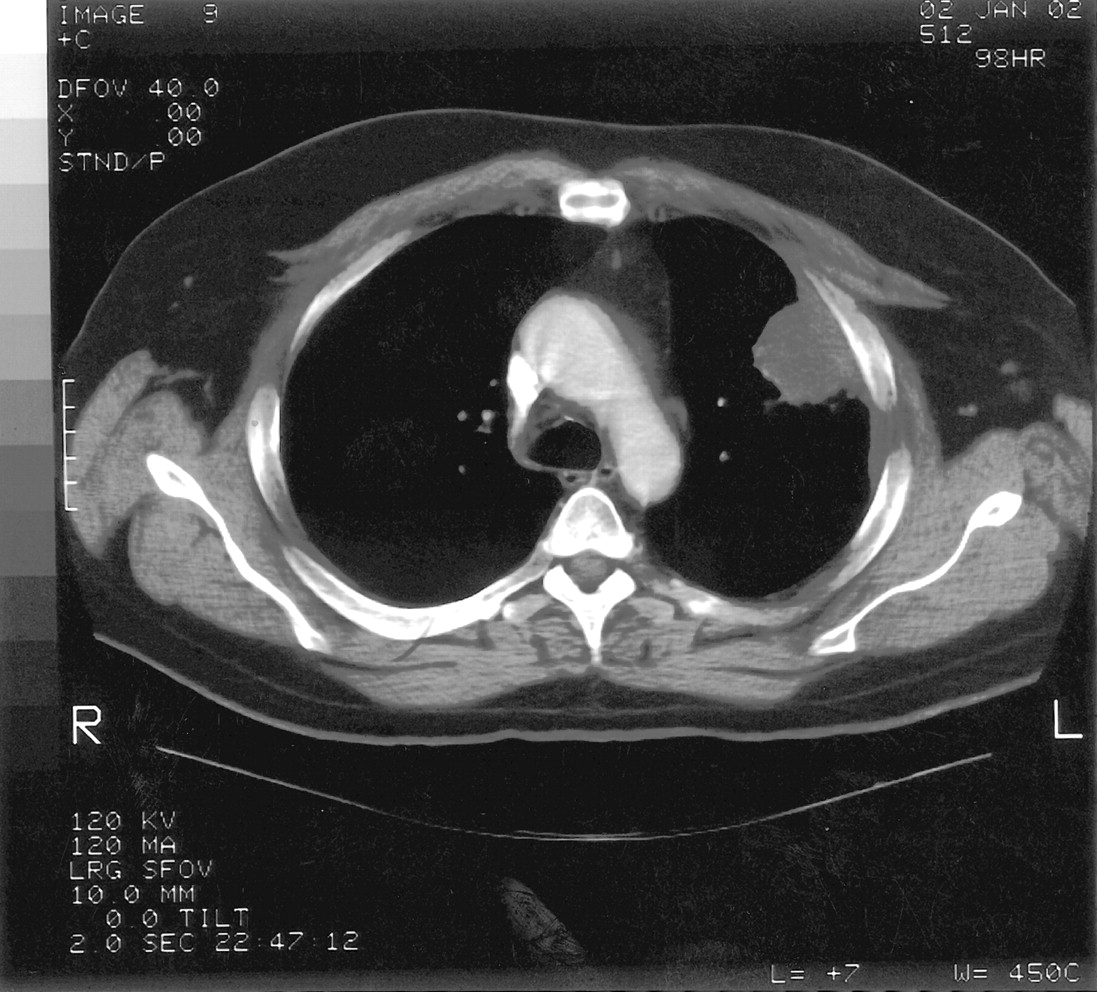
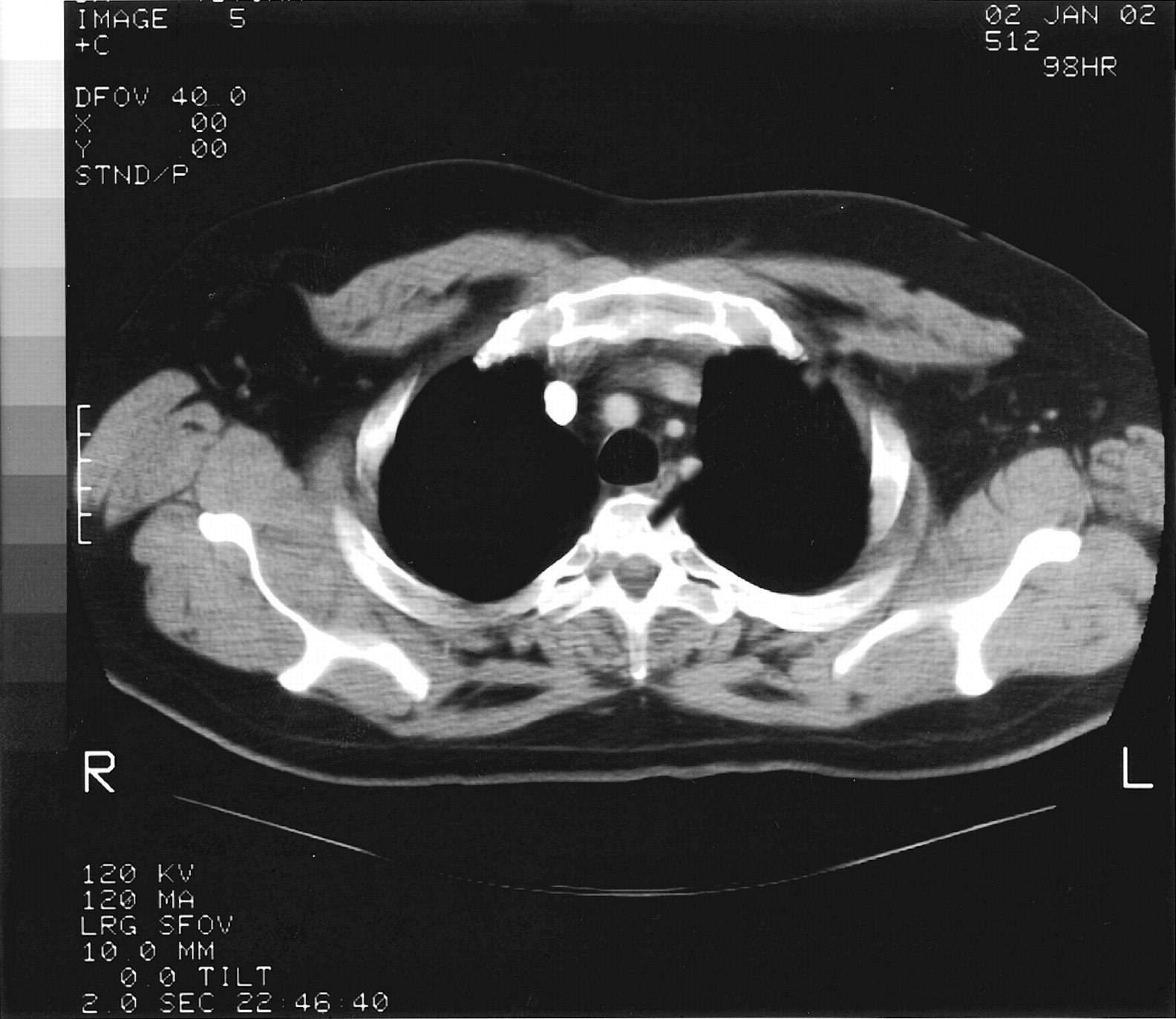
The admission diagnosis was pansinusitis that had failed outpatient treatment and a lung mass. The patient was started on 500 mg/day levofloxacin administered intravenously. Magnetic resonance imaging of brain ruled out possible cavernous sinus thrombosis. CT of the chest showed a large, dominant, left upper lobe lesion abutting the pleura with two smaller noncalcified lesions in the left and right upper lobes; this was interpreted as suspicious for carcinoma of the lung (Figs. 2 and 3). CT of his abdomen and pelvis were negative. Tumor markers (α-fetoprotein, prostate-specific antigen) were also negative.
Coronal CT of sinuses demonstrating bilateral ethmoid and maxillary sinus disease.
Chest CT demonstrating soft tissue mass in left upper lobe abutting the pleura.
On day 3, the patient complained of joint pain, mainly in the right shoulder and both hands. The patient’s erythrocyte sedimentation rate and C-reactive protein level were elevated. His rheumatoid factor level was 566 IU/mL, and tests for antinuclear antibodies were negative. He was started on indomethacin and his symptoms improved in 2 days. CT-guided biopsy of the lung showed acute and chronic interstitial inflammation suggestive of a granulomatous process (Fig. 4). The patient was put in respiratory isolation until the results of 3 sets of cultures for mycobacteria came back negative. Sputum cultures were negative for fungus.

Chest CT demonstrating small noncalcified lesions in the left upper lobe.
On day 10, the patient developed a painful ulcer on the left side of his tongue. Cultures were obtained from the lesion, and serum cytoplasmic antineutrophil cytoplasmic autoantibody (ANCA) levels were evaluated for WG. Results of the test for human immunodeficiency virus were negative. The patient’s symptoms improved significantly with indomethacin and levofloxacin. He was discharged from the hospital on day 13 taking levofloxacin, ibuprofen, and loratadine with a follow-up appointment in infectious disease clinic for culture result and evaluation of his lung mass. The patient was also scheduled for an appointment with the ear-nose-throat clinic. C-ANCA level was still pending on discharge.
Two days after discharge, the patient came back with difficulty in swallowing, flank pain, and purpuric rash on both of his lower extremities up to the abdomen. He was also complaining of dysuria and blood in the urine. His urinalysis showed positive nitrites, large amounts of blood, and more than 100 red blood cells per high-power field; 11 to 25 WBCs per high-power field, and moderate bacteria. Repeat erythrocyte sedimentation rate was 55 mm/hr, C-reactive protein level was 22.4 mg/dL, and rheumatoid factor was 1172.7 IU/mL. He had another painful ulcer on his uvula. The patient was admitted again with the diagnosis of Wegener granulomatosis, although ANCA levels were still pending from previous admission. He was started on 125 mg of intravenous methylprednisolone every 8 hours. A nephrologist was consulted.
Discussion
WG was first described by Klinger in 1933, followed by other investigators, including Rossle in 1933, Wegener in 1936 and 1939, and Ringertz in 1947.1
WG is currently characterized as one of the ANCA-associated small vessel vasculitides. It is distinguished clinically by its predilection for affecting the upper and lower respiratory tracts and kidneys and by the histologic presence of necrosis, granulomatous inflammation, and vasculitis.
There is a strong and specific association with autoantibodies directed against proteinase 3, a constituent of neutrophil azurophilic granules.2 The presence of such antibodies is a strong indicator for a diagnosis of WG, but should not be used in place of a tissue diagnosis. The presence of ANCA is not required to make a diagnosis of WG by either the American College of Rheumatology (ACR) or the Chapel Hill Consensus Conference (CHCC) definitions.3 Occasionally, patients with infections, neoplasms, inflammatory bowel disease, sclerosing cholangitis and other rheumatologic diseases develop ANCA, but these are predominantly perinuclear ANCAs or exhibit an atypical staining pattern.4
Most patients are white, the sex distribution is equal, and most present in the fifth decade, although the age range extends to both extremes.5 The clinical presentation of WG can be so diverse that the list of differential diagnoses is vast, ranging from infections (fungal, bacterial, and mycobacterial) to other vasculitides, including Henoch-Schönlein purpura, sarcoidosis, Behcet syndrome, and malignancies.6
Unexplained constitutional symptoms are often part of the initial presentation. Fever and weight loss may be reported at the onset of the disease and more frequently during the course of the illness.5 The upper airway disease is the most common presenting feature of WG. This includes sinusitis, oral lesions (ulcer, gingivitis), otitis media, hearing loss, epistaxis, and saddle nose deformity; sinusitis is the most frequent initial presentation in about half to two thirds of patients with WG.5,7
Pulmonary involvement is one of the cardinal features of WG. It occurs in 45% of patients at presentation and 87% during the course of the disease.5 Cough, hemoptysis, and pleuritis are the most common pulmonary symptoms. The most common radiographic findings include pulmonary infiltrate (67%) and nodules (58%).5
Renal disease also may be seen as the initial presentation or during the course of the disease. Once present, renal disease may progress from asymptomatic and mild to fulminant glomerulonephritis within days or weeks, resulting in end-stage renal failure.7,8 Even with appropriate therapy, it may lead to chronic renal insufficiency and renal failure.5
Ocular manifestations have been reported to occur in 28 to 58% of patients with WG, and they may be part of the initial presentation in 8 to 16% of patients.9,10 A complete ophthalmologic examination is an important part of the diagnostic evaluation. Any compartment of the eye may be affected. Keratitis, conjunctivitis, scleritis, episcleritis, nasolacrimal duct obstruction, uveitis, retro-orbital pseudotumor with proptosis, retinal vessel occlusion, and optic neuritis have all been described.5,9–11 Visual loss has been reported in as many as 8% of patients.5 CT or magnetic resonance imaging of the orbit and sinuses may provide useful anatomic information.
Other unusual presentations of WG include salivary gland, cutaneous, gastrointestinal, and cardiac involvement.5,7,8,11–18
Treatment
Traditionally, initial therapy of WG consists of daily oral cyclophosphamide-corticosteroid combination therapy. This treatment has been quite effective in inducing remission in more than 90% of patients who adhere to this regimen; approximately 75% experience complete remission.5,19,20 The mean time to complete remission is 12 months, with occasional patients requiring treatment for more than 2 years before all symptoms have resolved. Therefore, patients should not be considered nonresponders until they have been monitored on this regimen for more than several months. Response to this regimen is defined as a lessening or resolution of the inflammatory manifestations.19 This is important because new patients may have persistent abnormalities that are not caused by active disease. For example, patients in whom the systemic symptoms and signs resolve and their urinalyses become inactive are considered to be in remission, but they may have persistent or even slowly worsening renal insufficiency.
The use of aggressive immunotherapy in this disease is justified because survival in patients with untreated WG is extremely poor; up to 90% of patients die within 2 years, usually because of respiratory or renal failure.5,19,20 Mortality, however, can be significantly reduced with the introduction of a cyclophosphamide-corticosteroid therapy combination. The two routes of administration are oral and intravenous. Direct comparisons between oral and intravenous cyclophosphamide also have been performed in small prospective and randomized trials.20–23 Intravenous and oral cyclophosphamide have similar efficacy in terms of inducing remission.22,23 Intravenous therapy was associated with less toxicity but a trend toward a higher rate of relapse.23,24 In the medical literature, the daily oral cyclophosphamide and corticosteroid regimen is the preferred method of managing these patients. However, in patients who have limited or non–life-threatening disease, monthly intravenous cyclophosphamide pulse therapy can be effective and can minimize some of the side effects.21,22,24 Pulse therapy is given in conjunction with prednisone, usually starting at 1 mg/kg/day and given at this dosage for at least 1 month, then tapering by 5 to 10 mg per week until prednisone is stopped or the patient is placed on maintenance therapy of 5 to 15 mg every other day.
Other therapies for WG include methotrexate and prednisone, which is another alternative for patients with active but not immediately life-threatening disease and normal or near-normal renal function.25,26 This particular regimen may be attractive in patients who have limited bone marrow reserve from past cyclophosphamide therapy or a history of cyclophosphamide-induced bladder injury. Prednisone alone is not a recommended therapy for WG.5,24
In those with severe disease at presentation, pulmonary hemorrhage, or worsening disease despite immunosuppressive therapy plasmapheresis may be indicated.27,28 There is not much clinical data to address the long-term outcome in patients who undergo plasmapheresis for WG.29,30
In our case, the patient began therapy with monthly intravenous cyclophosphamide pulse therapy. The use of monthly intravenous pulse therapy is primarily to lower the overall cumulative dose of cyclophosphamide and thereby avoid some of the toxic side effects of the oral regimen. Our patient responded to intravenous pulse cyclophosphamide therapy and steroids with marked improvement in his urinalysis, decreased numbers of red blood cells per high power field, improvement in his serum creatinine level, and improvement of his inflammatory symptoms. He received a total of 6 months of intravenous pulse therapy, which he tolerated quite well, and has so far not had any signs of relapse.
- Revision received March 10, 2003.
References



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.