
Abstract
Pulmonary fibrosis is not uncommon. Usual interstitial pneumonitis (UIP)/idiopathic pulmonary fibrosis (IPF) is the most common of the idiopathic pulmonary fibrotic diseases and has the worst prognosis with a mean life expectancy of 3.8 years. The American Thoracic Society has provided guidelines for the accurate diagnosis of IPF.
In 2014, 2 antifibrotic medications were approved in the United States that target the multiple fibrotic pathways of UIP, which increased the need for early and accurate diagnosis of IPF. The early and correct diagnosis is hampered by mimickers that include nonspecific interstitial pneumonitis, chronic hypersensitivity pneumonitis, and fibrotic sarcoidosis. Careful history taking, serologic testing, and Computer Tomography (CT) inspection can frequently make the correct diagnosis without need of invasive procedure. The purpose of this article is to share the most important aspects of the clinical and radiology presentation of IPF and its mimickers to enhance primary care clinician's ability to correctly and noninvasively diagnose UIP/IPF.
Fibrosis is the final common pathway of many injuries to the lung. Perhaps the earliest known cause of fibrosis was inhaled antigen-mediated hypersensitivity pneumonitis. In 1713, Bernadino Ramazzini recorded the health hazards associated with 52 occupations. He detailed the breathing difficulties related to maple-bark mold causing hypersensitivity pneumonitis.1 In the early 1900s asbestos was touted as a fire-retardant material with excellent insulating capability. By the 1970s it was known to cause lung fibrosis and its usage was banned by the US Environmental Protection Agency. Radiation Fibrosis occurs in the lung when it is exposed to greater than 20 Gy of radiation.2 Osteophytes of the spine can cause pulmonary fibrosis from mechanical irritation.3
In 1969 Liebow and Carrington4 described a group of idiopathic interstitial pneumonias that included usual interstitial pneumonia (UIP) which is associated with the clinical diagnosis of idiopathic pulmonary fibrosis. The criteria for diagnosing UIP have been well established. Recently, the American Thoracic Society/European Respiratory Society provided an update of the Classification of Idiopathic Interstitial Pneumonias (IIPs). Four categories were defined: chronic-fibrosing IIPs, acute or subacute IIPs, smoking-related IIPs, and rare IIPs.5 In clinical practice the fibrosing IIPs, which include usual interstitial pneumonitis (UIP) and fibrotic nonspecific interstitial pneumonitis (NSIP), are most frequently encountered and provide the greatest diagnostic dilemma because of their overlapping clinical, radiologic, and pathologic presentation. Chronic hypersensitivity pneumonitis (CHP) and fibrotic sarcoidosis (S4), which are not listed as idiopathic fibrosis, further complicate diagnosis because of their relative frequency and similar presenting features. The goal of this review article is to provide an overview of the clinical and radiologic diagnosis of UIP/idiopathic pulmonary fibrosis (IPF) and mimickers of the disease with the main goal being ability to answer the question, “Is it IPF or not?” Differentiation of fibrosis is important because treatments are different as well as prognosis. IPF is treated with antifibrotic medications, NSIP and S4 are often treated with anti-inflammatory medications, and CHP requires removal of the antigen causing disease. Early disease diagnosis leads to improved outcomes for patients.
UIP/IPF
Clinical
UIP/IPF is classified as a fibrosing IIP and is the most common subtype of IIPs. UIP is the radiographic pattern and IPF is the clinical diagnosis associated with a UIP pattern. It is limited to the lungs and has the worst prognosis with median survival estimate of 3.8 years.6⇓–8 It is defined as a chronic fibrosing interstitial pneumonia of unknown cause with a UIP pattern on surgical lung biopsy or on high-resolution Computer Tomography (CT) scan (histologic UIP pattern: variegated pattern of alternating areas of normal or near-normal lung, juxtaposed to areas of lung remodeling with temporal heterogeneity of fibrosis consisting of scattered fibroblastic foci in the background of dense acellular collagen, and honeycombing).9 It frequently occurs in the elderly male population (median age, 66 years). Risk factors for disease include cigarette smoking and gastroesophageal disease.8 Most patients with IPF demonstrate a gradual worsening of lung function over years, whereas some patients experience episodes of acute respiratory worsening despite previous stability (ie, acute exacerbation).8 On physical examination the patients have crackles at the posterior lung bases. Pulmonary function tests demonstrate restrictive physiology with diminished Diffusion Capacity of Lungs for Carbon Monoxide (DLCO) and Forced Vital Capacity (FVC). According to the ATS/ERS/JRS/ALAT 2011 revised diagnostic criteria, the IPF diagnosis is securely established based on high-resolution computed tomography (HRCT) findings of UIP, and/or pathologic criteria, in the absence of known cause of lung fibrosis such as collagen vascular disease, drug toxicity, sarcoidosis, and various environmental exposures (i.e., CHP).8 In some patients with definitive HRCT findings (ie, UIP pattern; discussed in the following “Radiology” section), surgical lung biopsy can be avoided in the diagnosis of IPF. The multidisciplinary discussion among pulmonologists, radiologists, and pathologists experienced in the diagnosis of interstitial lung disease (ILD) increases the accuracy of the diagnosis. Most ILD centers in the United States have a dedicated ILD multidisciplinary conference, as vital part of the diagnosis and referring patients to those centers would expedite the diagnosis.10
Until recently, it had been believed that UIP/IPF was driven by uncontrolled inflammation, therefore many anti-inflammatory medications (prednisone, azathioprine, N-acetylcysteine, interferon-γ, etc.) were previously administered or tested in patients with UIP/IPF.11,12 However, recent evidence has demonstrated inefficacy or harm from these and they are not recommended therapy in the treatment of IPF.8 In the past decade, therapeutic modalities have been targeted at mechanisms involved in the wound healing cascade (ie, antifibrotic mechanism). In late 2014, 2 drugs, pirfenidone and nintedanib, were approved by the Food and Drug Administration (FDA) for treatment of IPF based on their ability to slow disease progression. Pirfenidone, an antifibrotic drug that reduces lung fibrosis through down-regulation of the production of growth factors and procollagens I and II, was shown to reduce the rate of FVC decline as well as a statistically significant improvement in progression-free survival in the Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND) trial11, which followed the PIPF004 and PIPF006 trials.13 Likewise, nintedanib, a tyrosine kinase inhibitor, originally developed as an antivascular agent for oncology indications, was tested for efficacy against IPF in several trials14,15, and these showed a significant impact in rate of FVC decline relative to placebo. These trials were not powered to answer effect on survival and given the lack of the data of long-term efficacy of these agents; UIP/IPF remains a refractory disease with guarded prognosis for which further investigation of new innovative therapies is necessary.
Radiology
Radiology plays an important role in the early and correct diagnosis of UIP. HRCT scans of the chest should be performed in the supine position in full inspiration. Intravenous contrast is not indicated. Slice thickness should be between 1 and 1.25 mm. Prone CT images can be acquired if the patient has early disease and there is a concern that the findings may represent dependent atelectasis. Expiratory images can be obtained on initial imaging if hypersensitivity pneumonitis is in the differential diagnosis because expiratory imaging demonstrates air trapping, one of the radiographic hallmarks of hypersensitivity pneumonitis.
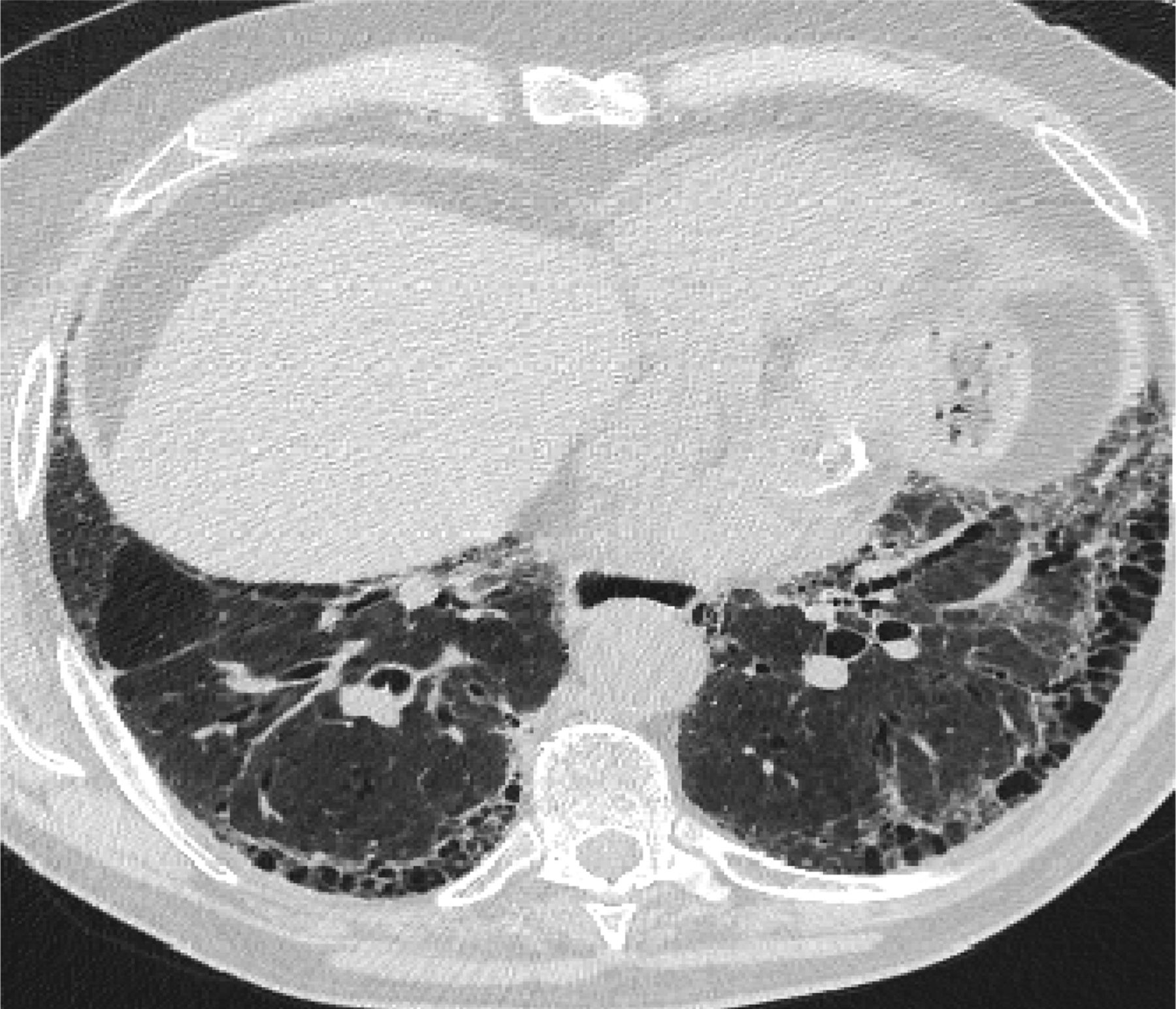
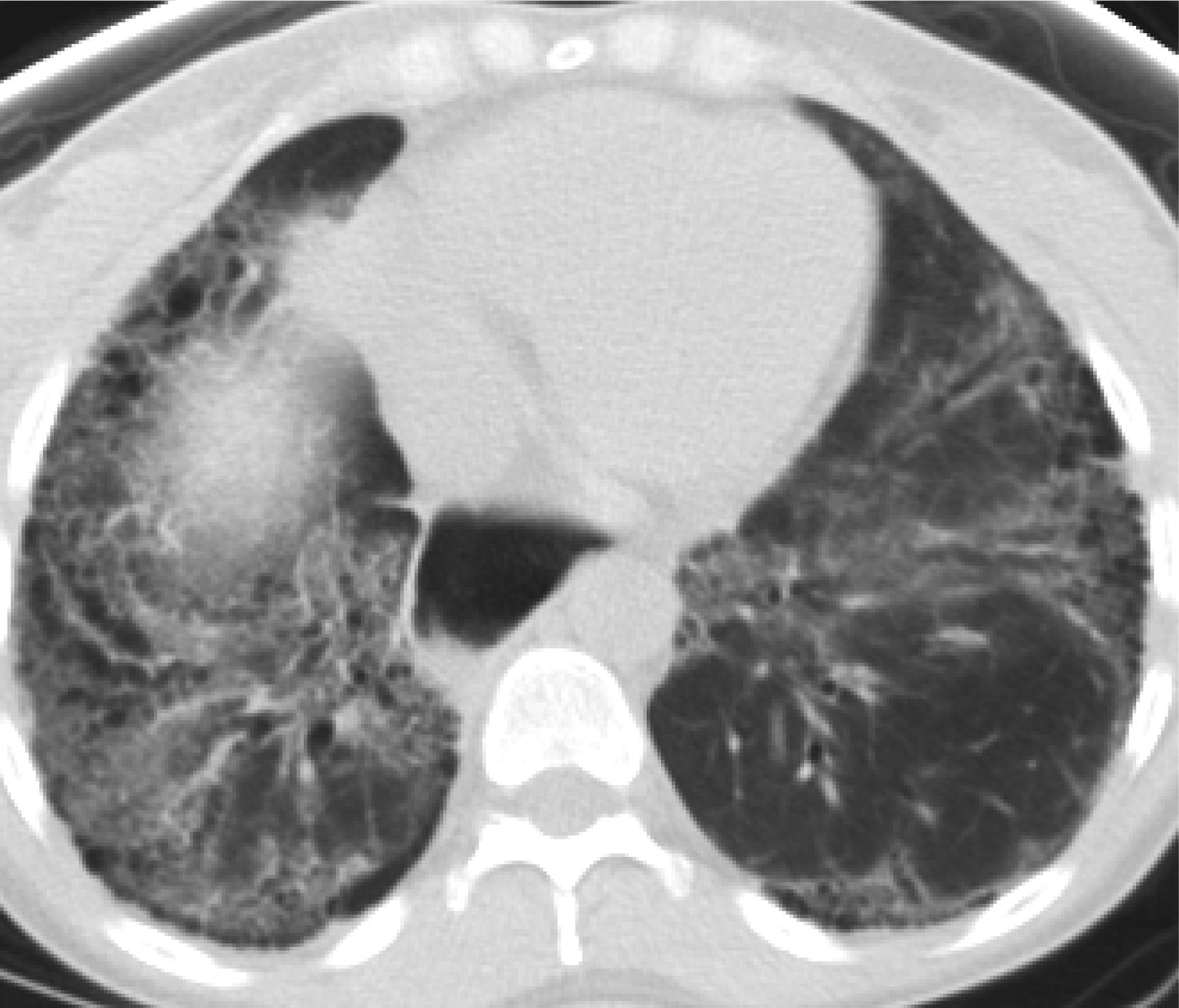
In 2011, the ATS/ERS/JRS/ALAT published evidence based guidelines for the diagnosis of IPF.8 A “UIP pattern” radiographically was defined as subpleural basilar predominant fibrosis, reticulations, honeycombing, and absence of features that would support an alternate diagnosis (Figure 1). The correct diagnosis of honeycombing is important and as such it deserves extra attention. The definition of honeycombing described by Webb et al16 is “rounded lucencies with shared walls in vertical stacks that are subpleural and occur in association with other findings of fibrosis.” The inter-reader agreement for honeycombing (HC) is low ranging, from 0.21 to 0.31 in previous reports17 due to mimickers of honeycombing, which include bronchiolectasis, paraseptal emphysema, and cystic bronchiectasis. If there is no honeycombing but other criteria are met, the diagnosis is a “possible UIP pattern” radiographically as per the ATS criteria (Figure 2). Recent articles support that the possible UIP pattern likely represents an early UIP pattern.18,19
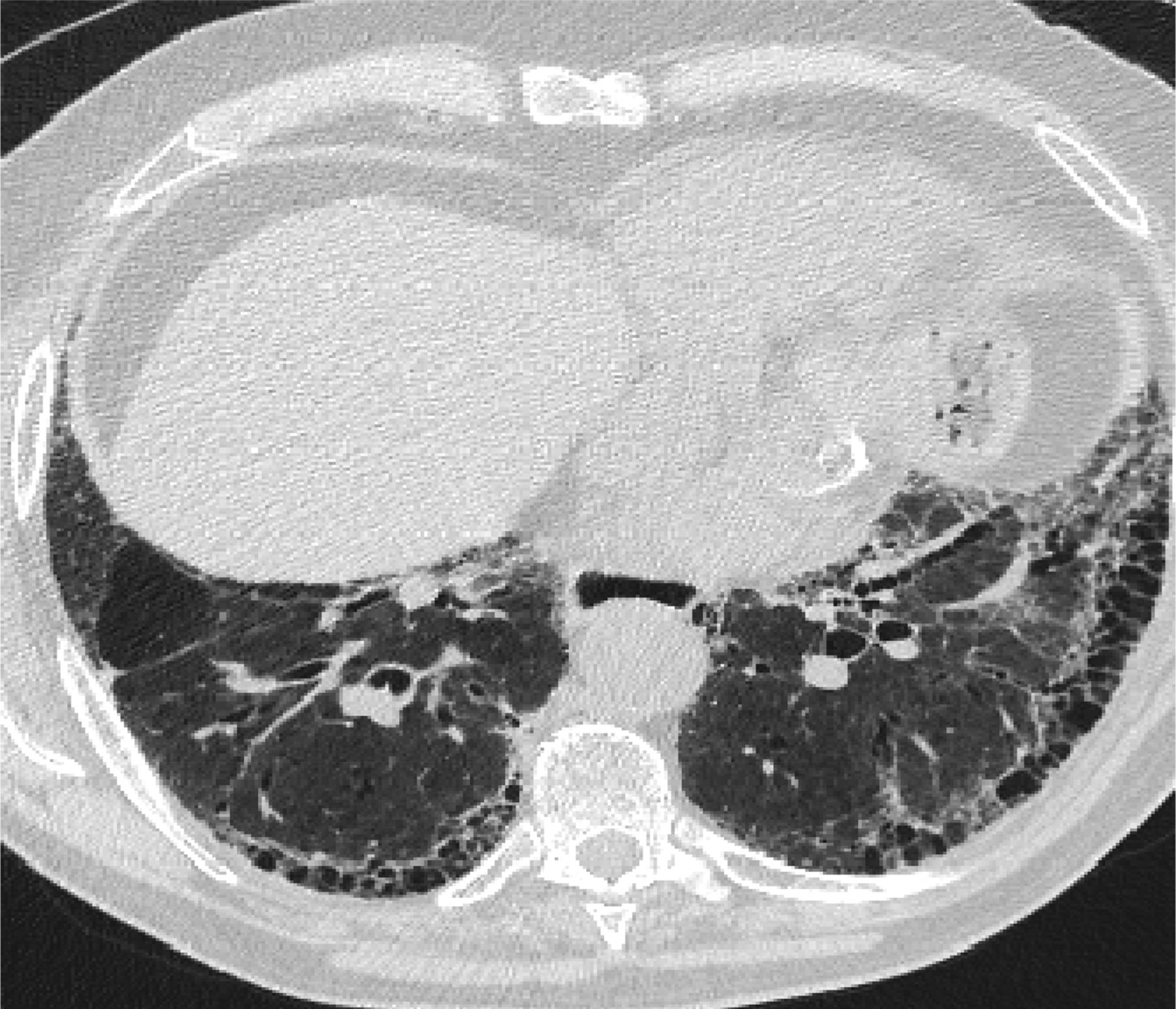
A “Usual interstitial pneumonitis (UIP) pattern” radiographically was defined by American Thoracic Society as subpleural basilar predominant fibrosis, reticulations, honeycombing, and absence of features that would support an alternate diagnosis.
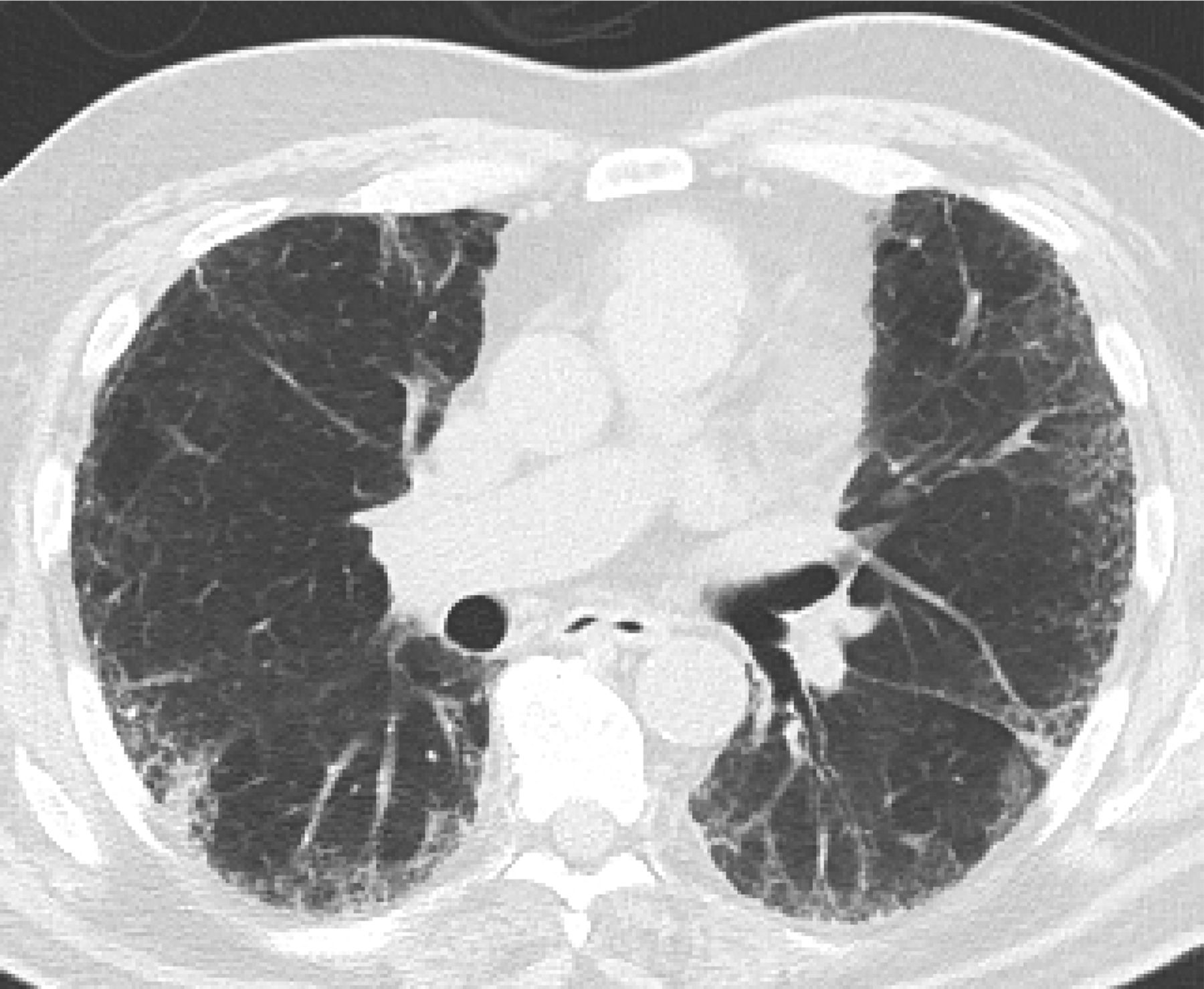
If there is no honeycombing but other criteria for a usual interstitial pneumonitis (UIP) pattern are met, the diagnosis is a “possible UIP pattern” radiographically as per the American Thoracic Society (ATS) criteria.
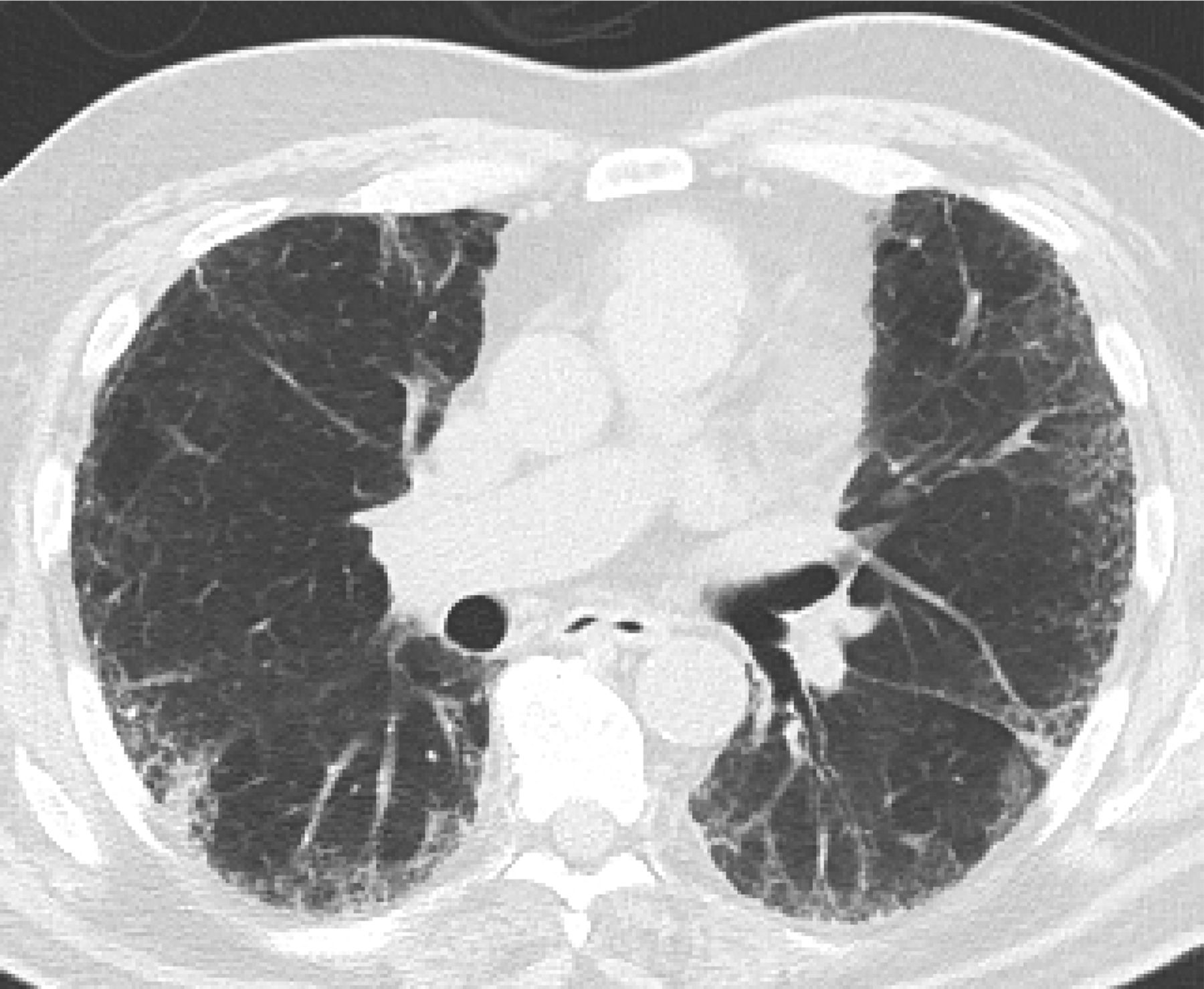
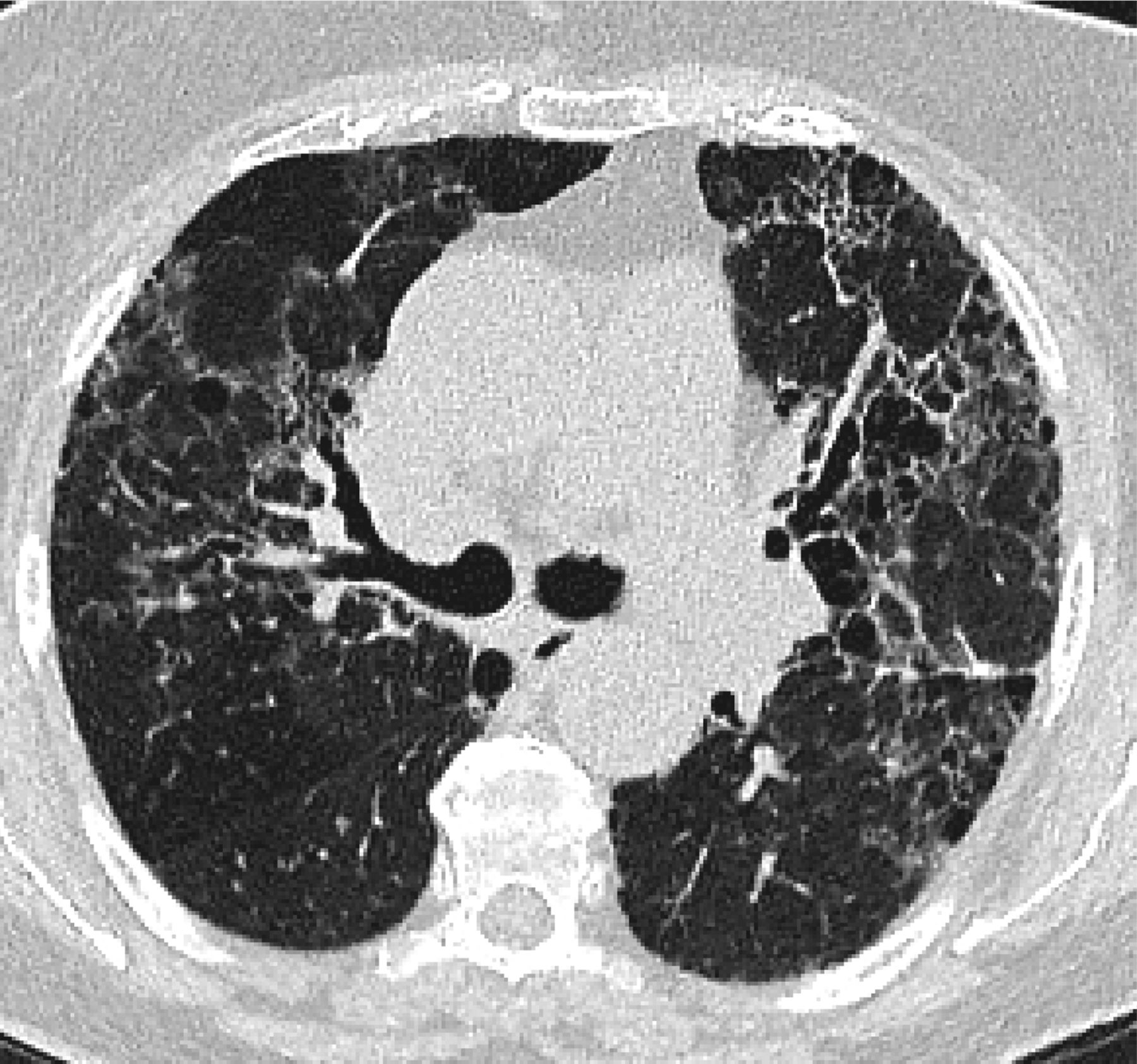
Features that would suggest an alternative diagnosis include consolidation as can be seen in organizing pneumonia (Figure 3), air trapping that is seen in hypersensitivity pneumonitis, and nodules, which are also seen in hypersensitivity pneumonitis and sarcoidosis. Ground glass opacities are frequently seen in patients with desquamative interstitial pneumonitis or respiratory bronchiolitis interstitial lung disease. Cysts occur in lymphocytic interstitial pneumonitis and lymphangioleimyomatosis among others. Bronchovascular distribution is consistent with hypersensitivity pneumonitis and NSIP and sarcoidosis. Upper lobe–predominant fibrosis is not typical of UIP and is more common with hypersensitivity pneumonitis and sarcoidosis.8

Features that would suggest an alternative diagnosis include consolidation as can be seen in organizing pneumonia, which follows the bronchovascular bundles in this example.
Mimicker No. 1: Fibrotic NSIP
Clinical
NSIP is a subtype of fibrosing IIPs and the first mimicker of UIP/IPF. In 1990s, it was reported that a subset of patients diagnosed with IPF had cellular infiltration on lung biopsy, bronchoalveolar lavage lymphocytosis, and better clinical response to anti-inflammatory therapy (ie, steroid, etc.) with a favorable long-term prognosis.20⇓⇓–23 NSIP histopathologic pattern is characterized by varying degree of inflammation and fibrosis with temporal uniformity (ie, varying proportions of interstitial inflammation and fibrosis seem to have occurred over a single time span, distinct from the temporal heterogeneity observed in UIP pattern). NSIP is most common among women in their 40s to 50s and nonsmokers, in contrast with UIP/IPF. NSIP is the most common histologic finding in some forms of connective tissue disease–related ILD (CTD-ILD) which usually does not require invasive diagnostic modalities such as surgical lung biopsy for diagnosis.24⇓–26 Therefore, specific attention should be given to connective tissue symptoms and signs (arthralgias, arthritis, skin changes, esophageal abnormalities, fever, etc.).Obtaining a comprehensive panel of serum autoantibodies and inflammatory markers, including but not limited to antinuclear antibody, rheumatoid factor, anti-Scl-70, antisynthetase antibodies (myositis panel), anti-Ro (SS-A), anti-La (SS-B), antiribonucleoprotein, aldolase, creatine kinase, erythrocyte sedimentation rate, anticyclic citrullinated peptide, and C-reactive protein, is crucial (Table 1). Many patients present with an underlying autoimmune feature but do not meet established criteria for a CTD; therefore, ERS/ATS recently proposed the term, “interstitial pneumonia with autoimmune features” (IPAF) and offered its diagnostic criteria.27 This new guideline suggests a possible underlying autoimmune etiology in patients with NSIP despite a lack of definitive CTD diagnosis. Moreover, since CHP and NSIP have many overlapping findings on radiography and histopathologic examination, a comprehensive environmental, occupational, and avocational history is also a critical step. After exclusion of UIP/IPF, CHP, and CTD, a clinician must confirm the histologic diagnosis obtained either by surgical lung biopsy or bronchoscopic cryobiopsy since definitive diagnosis of NSIP can only be made histologically.28 The majority of cases are typically classified as fibrosing, with less than 20% deemed to be cellular.29 As is the case with diagnosing UIP/IPF, the multidisciplinary discussion plays a key role in accurate diagnosis of NSIP and referring to ILD centers would be expected.
Serologies and Their Implications in Workup of Lung Fibrosis
In mild or asymptomatic diseases, serial monitoring of symptom and pulmonary function tests are sufficient. If progression of disease is seen, treatment with immunosuppressants is thought to be beneficial.30,31 Steroids are the predominant agent of choice, whereas other immunosuppressants (azathioprine, cyclophosphamide, cyclosporine, and mycophenolate mofetil) are used to supplant steroid therapy. The prognosis for NSIP is generally favorable compared with UIP/IPF, although there is an approximate 20% mortality rate in 5 years.24
Radiology
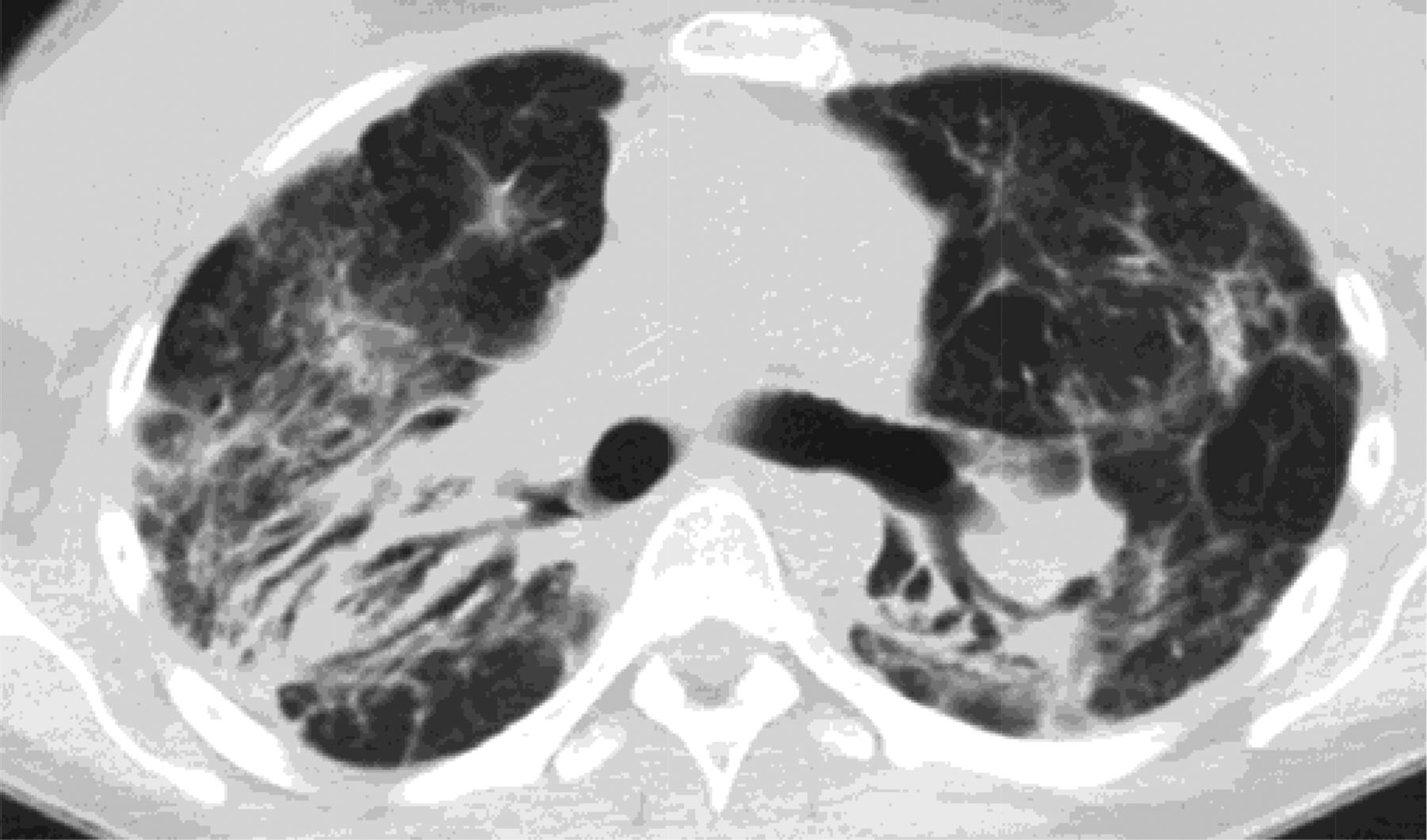
NSIP is a lower lobe–predominant fibrosis like UIP. However, in contrast with UIP, which is heterogeneous, NSIP is homogeneous.32 It also differs from UIP in that it is not subpleural but instead follows the bronchovascular bundles and in fact often spares the exact periphery of the lung.33 There are basically 3 types of NSIP radiographically, cellular, fibrotic, and mixed. Cellular NSIP is associated with ground glass opacities and has minimal volume loss. Cellular NSIP is more likely to respond to steroid treatment (Figure 4).

Cellular nonspecific interstitial pneumonitis (NSIP) is associated with ground glass opacities and has minimal volume loss. Cellular NSIP is more likely to respond to steroid treatment.
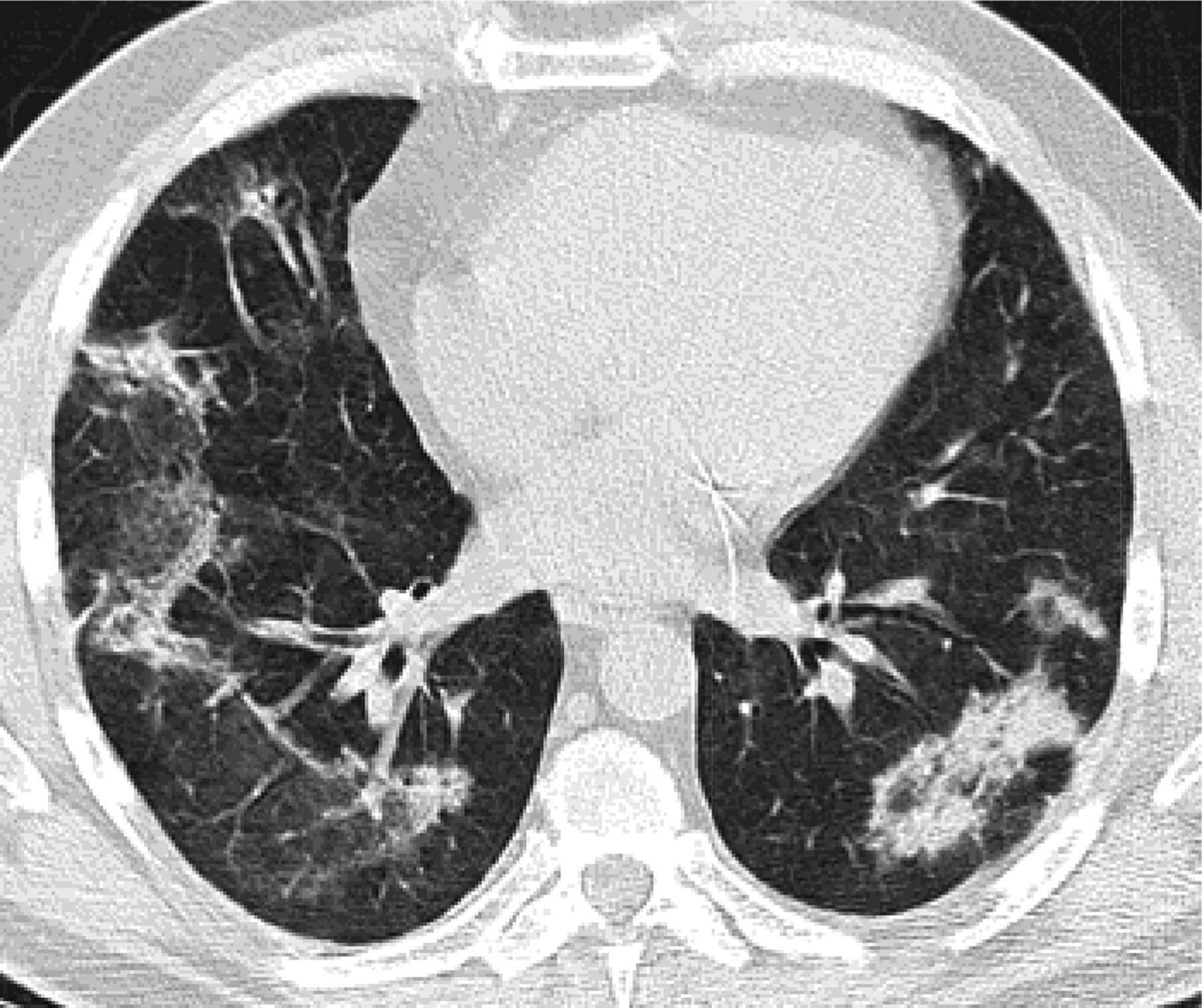
Fibrotic NSIP has less ground glass opacity and more volume loss displacing the major fissures posteriorly (Figure 5). This pattern can be confused with UIP. Fibrotic NSIP is less likely to respond to steroid treatment. In reality most cases of NSIP are mixed cellular and fibrotic with ground glass opacity and volume loss.

Fibrotic nonspecific interstitial pneumonitis (NSIP) has less ground glass opacity and more volume loss then cellular NSIP. The dilated esophagus in this photograph points to the cause of fibrosis, which was scleroderma.
Differentiation Pearl
NSIP and UIP are both lower lobe–predominant diseases; however, NSIP is distinctively different radiographically from UIP because of its homogeneity and its subpleural sparing.
IPAF presents most common radiographically as a lower lobe–predominant fibrosis that follows the bronchovacular bundles, but unlike NSIP it is more heterogeneous.
Mimicker No. 2: CHP
Clinical
CHP is an interstitial lung disease in genetically predisposed individuals caused by an exaggerated immune response to chronic inhalation of a variety of antigens in the environment (fungal, bacterial, protozoal, and animal proteins, or low-molecular-weight chemical compounds, etc.). With long-term inflammation, CHP with progressive fibrosis and bronchiolitis obliterans may develop and fibrosis is often characterized by honeycombing so that in late chronic stages, histopathology may be similar to UIP or fibrotic NSIP pattern. Therefore, CHP is an important mimicker of UIP/IPF.34⇓⇓⇓⇓–39 CHP frequently occurs in the elderly female population. However, the accurate prevalence or incidence of CHP is difficult to evaluate given that the disease is oftentimes unrecognized or misdiagnosed and exposure conditions vary in intensity of exposure (usually low) and from place to place, and country to country. The onset of disease is insidious with gradually increasing dyspnea on exertion, dry cough, fatigue, and weight loss. Given that patients seldom relate their symptoms to the environmental exposure and the onset of respiratory symptoms is gradual, physicians often misdiagnose the disease for another interstitial lung disease such as UIP/IPF. Diagnosis should be suspected in every patient with insidious respiratory symptoms A careful history regarding the occupational/domestic environment and hobbies is crucial (ie, bird keeping, hay feeding, feather duvet and pillows at home, air conditioning, contaminated ventilators in the buildings, and formation of mold on room walls or in brake fluid oils, or within wind instruments). Proof of sensitization (serum precipitins) and demonstration of a consistent pattern of this ILD on HRCT (discussed in the following “Radiology” section) support the accurate diagnosis. In contrast, pathologic diagnosis is not often required, but when obtained may demonstrate features associated with this disease even in cases with a UIP pattern (bonchocentric inflammation, lymphohystiocitic cell and poorly formed granulomas). Invasive lung biopsy may be avoided with careful history taking and radiologic evaluation by HRCT.
The prognosis of CHP varies among patients and depends on the duration of exposure to the inhaled antigen. Despite a favorable prognosis in the acute and subacute forms, CHP may become a progressive fibrotic lung disorder that results in respiratory failure even after avoiding the presumed antigen and the institution of therapy. In addition to avoidance to further antigen exposure, steroid therapy is usually recommended if patients show progressive functional impairment and immunosuppressants can be added as steroid-sparing agents.35
Radiology
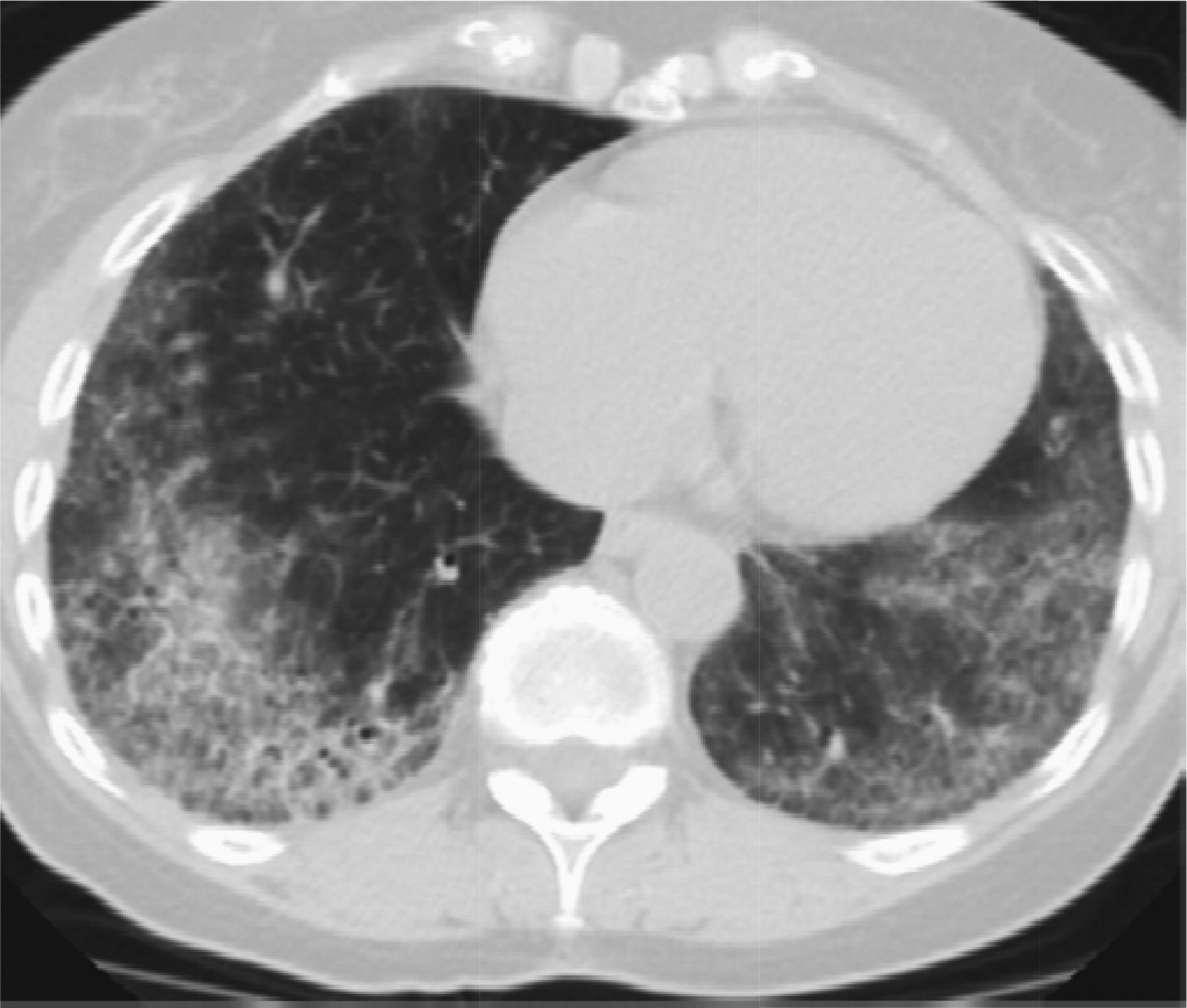
Hypersensitivity pneumonitis can be divided into acute, subacute, and chronic forms. The chronic form is most likely to mimic UIP radiographically. CHP is frequently misdiagnosed as IPF, which is particularly distressing because early recognition of disease and removal from antigen allows for cure.43 CHP is different from UIP radiographically mainly because instead of being peripheral, it is an airway-centered disease, which makes sense given the way in which it is acquired. Second, CHP is upper-lobe predominant and unlike UIP, which is lower-lobe predominant (Figure 6). Third, CHP frequently demonstrates air trapping on expiratory CT images.41 Air trapping is infrequently associated with a UIP pattern and when present a diagnosis of rheumatoid arthritis should be considered.42

Chronic hypersensitivity pneumonitis (CHP) is upper-lobe predominant, airway centered, and frequently has air trapping. The most useful feature radiographically is its airway-centered distribution seen on this image.
Differentiation Pearl
CHP is heterogeneous like UIP but its bronchovascular distribution makes it uniquely different from UIP. Air trapping is also important in making the correct diagnosis of CHP and is rarely seen in association with UIP.
Emphysema is frequently seen in association with UIP but rarely with CHP.
Mimicker No. 3: Stage 4 Sarcoidosis
Clinical
Sarcoidosis is a multisystemic inflammatory disease of unknown etiology, characterized by the presence of noncaseating granulomas, and predominantly affecting lung. In the majority of patients, pulmonary sarcoidosis undergoes clinical remission either spontaneously or with therapy and favorable long-term outcomes are achieved. However, approximately 20% of patients develop pulmonary fibrosis (ie, radiographic stage iv sarcoidosis) with substantially increased mortality, therefore it can be another mimicker of UIP/IPF.43,44 Stage 4 Sarcoidosis is a fibroticdisease with little or no granulomatous inflammation and clinical improvement is not expected with anti-inflammatory therapy.45 Fibrosis in sarcoidosis originates from granuloma and along with bronchovascular bundles may result in bronchial distortion and large cystic changes, and interlobular septal fibrosis results in linear scarring.46 The histologic features of UIP pattern (honeycombing, fibroblast foci, etc) are not typical in sarcoidosis. In sarcoidosis, wheezing, which is attributed to airway-centric fibrosis is common, although patients are less symptomatic than UIP/IPF.47,48 In contrast with IPF, acute exacerbations of disease attributed to diffuse alveolar damage have not been reported in stage 4 sarcoidosis.43
Treatment is indicated in patients who are symptomatic, with progressively worsening pulmonary function.44 Pulmonary fibrosis is an irreversible event, but in at least some patients, fibrosis coexists with active granulomatous inflammation. This is often difficult to discern and tests that suggest activity such as Gallium or Positron Emission Tomography (PET) scan are utilized to guide therapy. Treatments include multiple modalities aimed at suppressing inflammation with the use of anti-inflammatory agents (corticosteroids, methotrexate, azathioprine, leflunomide, mycophenolate mofetil, TNF antagonist, etc.). Although life expectancy is longer in S4 compared with UIP/IPF, once patients develop end-stage fibrotic lung disease, survival is limited and lung transplantation may be the treatment of last resort as it is for selected patients with pulmonary UIP/IPF.43
Radiology
Stage 1 sarcoidosis demonstrates hilar and mediastinal lymphadenopathy, stage 2 sarcoidosis manifests as adenopathy and pulmonary nodules or densities in a peri-lymphatic distribution, stage 3 sarcoidosis has parenchymal involvement without lymphadenopathy, and stage 4 disease presents with fibrosis of the lung, which needs to be differentiated from UIP and CHP (Figure 7). Stage 4 sarcoid is an upper lobe–predominant fibrosis, which helps to differentiate it from UIP. In addition, it is not peripheral but instead is airway centered. Unlike CHP it tends to be posterior in the upper lobe and there is no air trapping.49,50
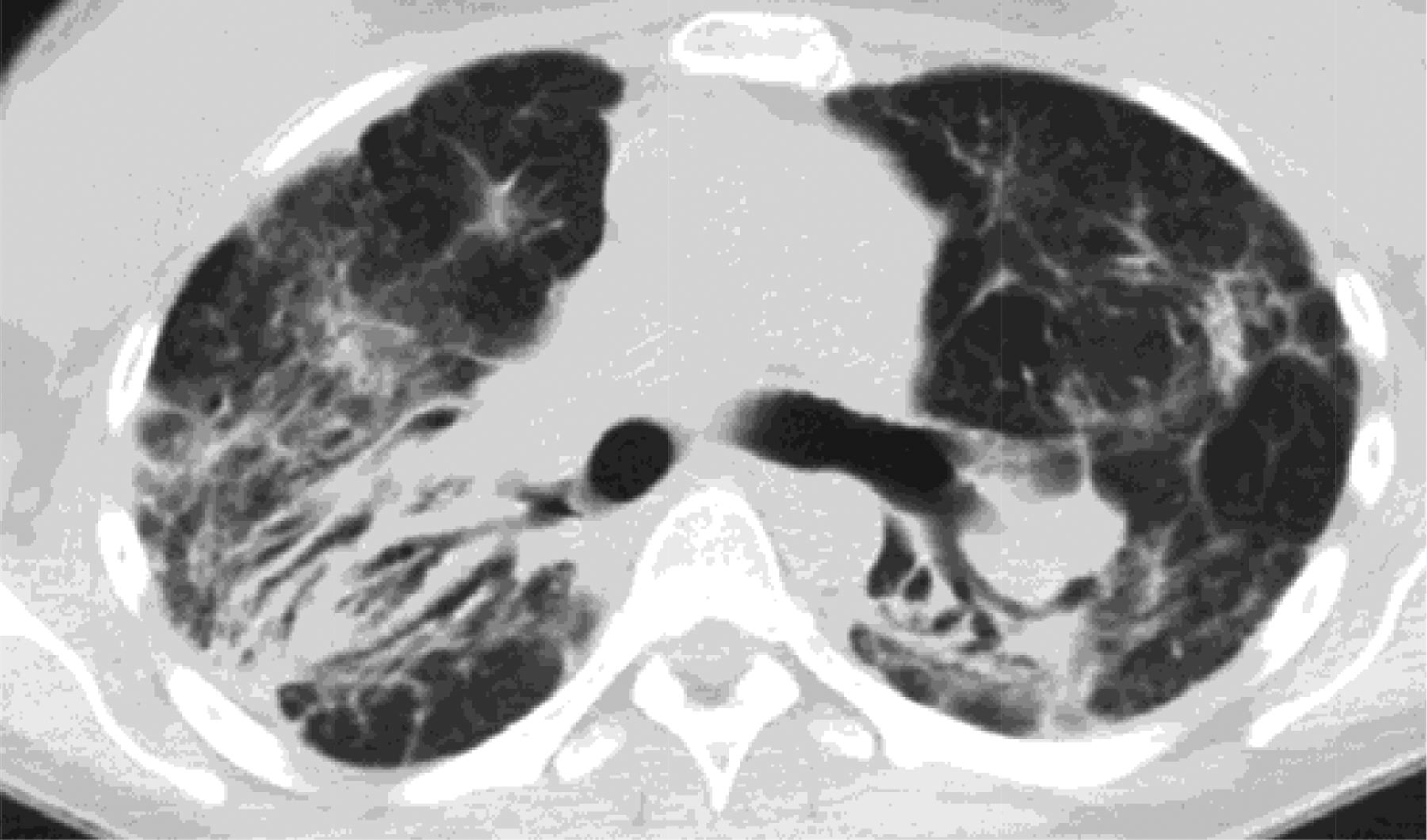
Stage 4 sarcoidosis is an upper-lobe predominant fibrosis, which helps to differentiate it from usual interstitial pneumonitis (UIP). In addition, it is not peripheral but instead is airway centered.
Differentiation Pearl
Sarcoidosis has more in common radiographically with NSIP and CHP than with UIP because the fibrosis is not peripheral but instead follows the bronchovascular bundles.
Its upper-lobe predominance helps to differentiate S4 from NSIP.
A posterior predominance and absence of air trapping helps to differentiate S4 from CHP, which is upper lobe but more frequently anterior.
Conclusion
UIP/IPF has a poor prognosis with a mean life expectancy of 3.8 years. The American Thoracic Society (ATS) has provided guidelines for the accurate diagnosis of IPF. This is a diagnosis of exclusion. There must be no known cause for a patient's lung fibrosis. The CT scan must show a UIP pattern with subpleural basilar-predominant fibrosis and honeycombing and absence of features that would suggest alternative diagnoses. A “possible UIP pattern” by ATS criteria includes the same criteria except honeycombing. If the patient does not have HC a lung biopsy may be considered to make the definitive diagnosis.8
In 2014, 2 antifibrotic medications, nintedanib and pirfenidone, were approved in the United States for the treatment of IPF. These daily administered medications are expensive but are often covered by insurance if patient has a diagnosis of UIP/IPF. These target multiple pathways of UIP51 and slow the decline of the forced vital capacity (FVC). They may be helpful in other fibrotic lung disease but his has not been studied. Therefore, it is now critical to diagnose IPF early and accurately. Early referral to a pulmonologist with expertise in lung fibrosis may be beneficial. The early and correct diagnosis is challenged by mimickers which have similarities to UIP/IPF and include NSIP, CHP, and sarcoidosis. Fortunately, careful clinical history, serologic testing, CT inspection, and multidisciplinary discussion can establish the correct diagnosis without an invasive procedure (Tables 2 and 3). It has been the goal of this article to share with primary care clinicians the most important aspects of the clinical and radiology presentation of IPF and its mimickers so that you will be able to correctly and noninvasively diagnosis UIP/IPF.
Overview of Common Fibrotic Lung Diseases
Putting It All Together: Typical Scenarios for Patients with Lung Fibrosis
Notes
This article was externally peer reviewed.
Funding: none.
Conflict of interest: Dr. Salvatore is a lecturer at Genentech, Boehringer Ingelheim, and Rockpointe.
To see this article online, please go to: http://jabfm.org/content/31/1/151.full.
- Received for publication July 7, 2017.
- Revision received September 22, 2017.
- Accepted for publication September 26, 2017.
References
In this issue



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Content Usage and the Most Frequently Read Articles of 2018
- Healthcare utilisation and costs in the diagnosis and treatment of progressive-fibrosing interstitial lung diseases
- Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases
- Interventions Must Be Realistic to Be Useful and Completed in Family Medicine