
Abstract
Long QT syndrome is a potentially lethal cardiac channelopathy that can be mistaken for epilepsy in young people. We report a 17-year-old man who was initially treated as having both daytime and nocturnal idiopathic epilepsy for 5 years. A series of electrocardiograms showed the time of the convulsive episodes, and genetic testing lead to the final diagnosis. The combined use of a β-blocker and a pacemaker implant incompletely abolished the torsade de pointes. After an additional near-fatal event, a cardioverter defibrillator was implanted as final bridge therapy. An electrocardiogram with the correct calculation of the QT interval should be performed on all young people with a suggestive history; that is, treat refractory convulsive episodes specifically with nondiagnostic electroencephalograms.
Congenital long QT (LQT) syndrome, one of the leading causes of sudden cardiac death in people younger than 20 years of age and is a potentially lethal cardiac channelopathy that can be mistaken for epilepsy. Disease prevalence is estimated to be anywhere between 1 in 5,0001 to 1 in 20,000.2 The mortality rate in untreated patients with congenital LQT syndrome is in the range of 1% to 2% per year3 and 20% within the first year of diagnosis.4 The consequences of untreated or mistreated LQT syndrome can be disastrous. Some syndrome subtypes may result in convulsions during sleep or rest that are often incorrectly attributed to a seizure disorder, resulting in inappropriate treatment with anticonvulsants. Here we present a teenager with congenital LQT syndrome who, for 5 years, had recurrent convulsions that had been treated as epilepsy. LQT syndrome should be considered and a series of electrocardiograms (ECGs) should be obtained in any young patient presenting with “resistant epilepsy,” particularly with nondiagnostic electroencephalograms (EEGs).
Case Report
A 17-year-old man was hospitalized for repeated convulsions. At home he had tonic-clonic convulsions associated with upward gaze. These episodes usually lasted for 1 to 2 minutes. He had full recovery shortly after the episodes without any significant neurological deficits. These convulsions occurred approximately 4 to 5 times per year, beginning at age 12. The episodes usually occurred at sleep and were often associated with urinary incontinence. He had been diagnosed as having idiopathic epilepsy by a number of pediatric neurologists and was being empirically treated with anticonvulants (carbamazepine, valproic acid, and phenytoin). During physical examination he was alert and well developed. Electrolyte levels, including potassium, magnesium, and calcium, were within normal range. Magnetic resonance imaging of the head was normal. Neurologic findings, including hearing function, were also normal. An EEG was obtained a day later and was normal when the patient was both awake and asleep. General cardiac examination, echocardiogram, and coronary arteriography were unremarkable. There was no history of seizures, syncope, or sudden death in any family members or close relatives. The 12-lead ECG of his parents and sibling were unremarkable, showing QTc <420 ms for each.
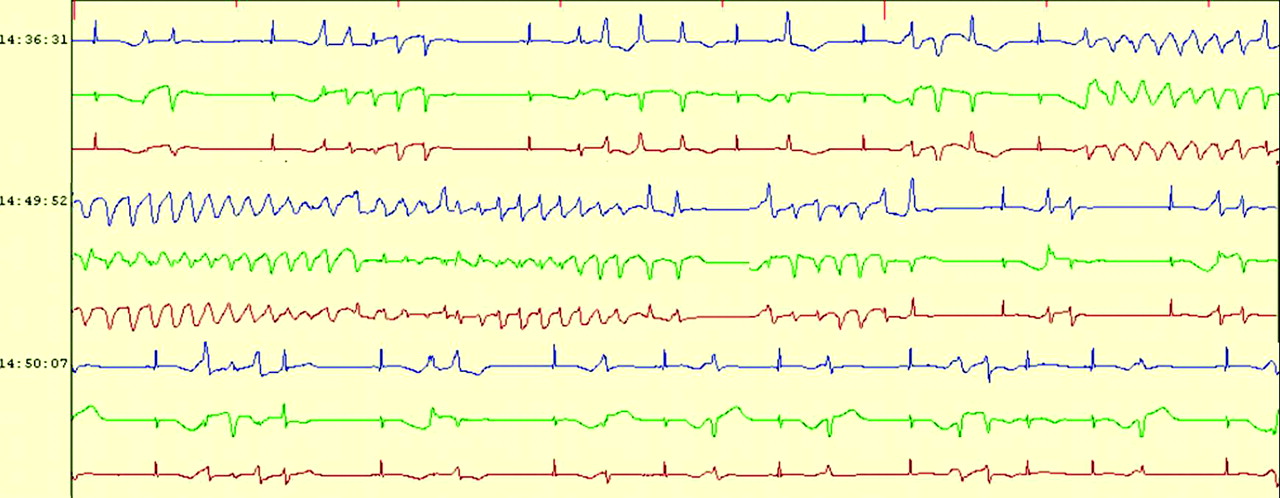
During his hospital stay, a generalized tonic-clonic seizure during sleep with spontaneous recovery was witnessed. A rhythm strip obtained immediately afterward showed sinus pause, bifid T wave, and T wave alternans (Figure 1). A trademark dysrhythmia of unsustained torsade de pointes (TdP) with a QTc duration of 518 ms (Figure 2) was recorded shortly after further cardiac consultation was begun. A Holter recording obtained during another ictal episode documented frequent runs of self-terminating polymorphic ventricular tachycardia and TdP (Figure 3), ultimately leading to the final diagnosis. Genetic testing revealed a mutation of the human ether-a-go-go-related gene (HERG or KCNH2) (a deletion of one base pair in exon 12, 2768delC) encoding the rapidly activating delayed rectifier cardiac potassium channel (IKr), a finding consistent with LQT2 syndrome. Genetic analysis for the patient's family members and other relatives was negative. LQT syndrome was diagnosed based on the “Schwartz and Moss” clinical criteria,5 including the recording of a low heart rate for his age, T wave alternans, notched T waves, prolonged QTc, and TdP. After informed discussion he decided against having an implanted cardioverter defibrillator (ICD) because of a lack of financial support; thus a pacemaker was implanted at that time. The antiepileptic drugs were discontinued and he was started on a course of propranolol and spironolactone. After that, the patient was free of seizure episodes until sudden cardiovascular collapse occurred 74 months later while he was eating a meal. On arrival at the emergency department he was pulseless and apneic, and an ECG showed ventricular fibrillation. Defibrillation was required twice before sinus rhythm and gasping respiration returned. An ICD was placed and he was discharged 1 week later. He continued taking propranolol and spironolactone. He returned to have his job as usual and was still asymptomatic (without ICD shock) 38 months after the intervention.
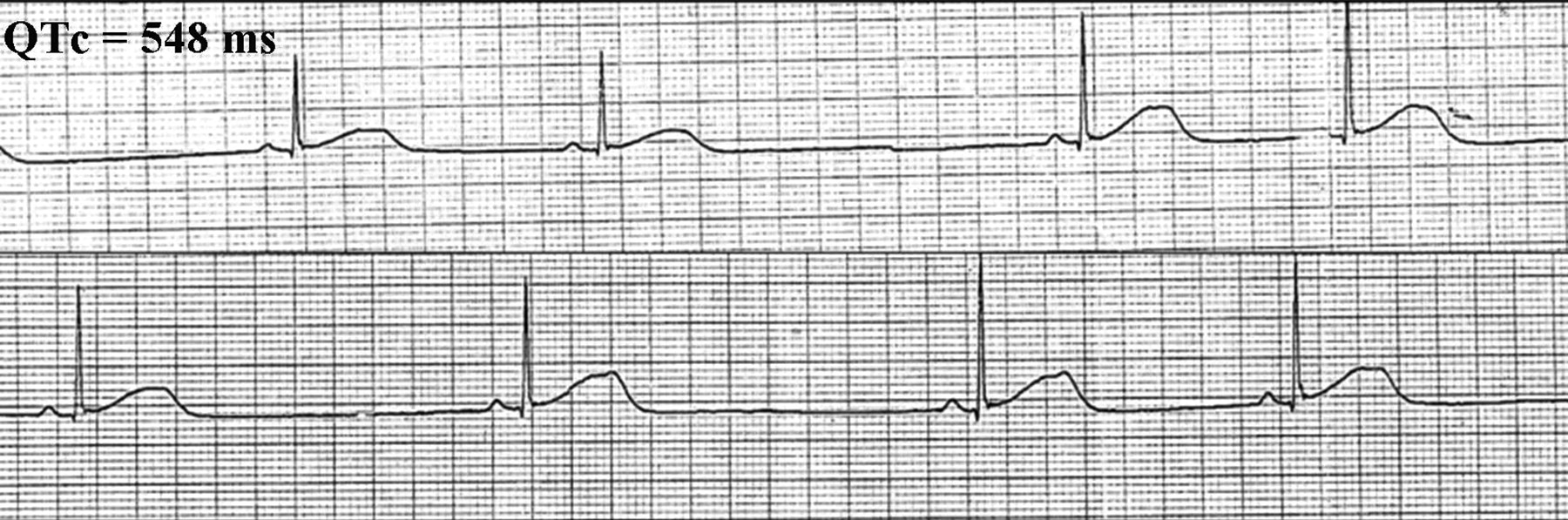
Rhythm strip demonstrating prolonged QTc; bifid T wave, a hallmark of the LQT2 genotype; and T wave alternans. LQT2, congenital long QT syndrome subtype 2.

A 12-lead ECG showing an unsustained torsade de pointes with a QTc duration of 518 ms was recorded shortly after a seizure episode. QTc, corrected QT interval (measured QT/square root of RR in seconds).

Frequent runs of self-terminating polymorphic ventricular tachycardia and torsade de pointes were detected on Holter monitoring during an ictal episode.
Discussion
Inherited cardiac ion channel abnormalities can cause convulsive activity and sudden death as a result of ventricular arrhythmias in the absence of structural heart disease. Affected subjects with LQT syndrome often “fly under the radar,” with the disease remaining undiagnosed or untreated for years. In this case, 5 years elapsed between the first seizure and the correct diagnosis of LQT syndrome. With the potential for fatal consequences in undiagnosed individuals, the early recognition of LQT syndrome is of paramount importance. These patients have a primary cardiac problem, and most often the self-terminating TdP and resultant hypoxemia result in a convulsive episode. In a small minority of events the TdP degenerates into ventricular fibrillation and sudden death occurs. If the attention in such cases is focused on the convulsive activity that may follow the patient may be mistreated because it is assumed to be epilepsy. Indeed, the diagnosis of LQT syndrome primarily depends on a high index of suspicion and the ECG findings. Standard ECG recordings and analysis of QTc duration and T wave morphology are the most useful tests. However, it must be emphasized that not all cases of LQT syndrome can be easily diagnosed by a single ECG lead. The QTc may vary, and prolonged QT interval may not be evident in all ECG leads. Even in known genetic carriers, the QTc at rest may be normal or borderline but prolonged at other times, particularly with β-adrenergic stimulation or during exercise.6,7 Thus, a series of ECG monitoring (particularly EEG/ECG-video recording during an ictal event) may help make the diagnosis. A genetic defect in both cardiac potassium (LQT1 and LQT2) or sodium (LQT3) channels results in prolonged cardiac repolarization and TdP but the channels differ in the triggering factors for attacks.8 Clinical characteristics in common genotypes of congenital LQT syndrome are summarized in Table 1. In the LQT1, most life-threatening cardiac events occur in association with physical or emotional stress. However, LQT3 patients are more likely to have TdP at rest or during sleep (during either the day or night) without arousal. In patients with LQT2, triggers include both rest and exercise, as well as loud noises, like being awakened by an alarm clock.9 So far, at least 10 different genes and >200 mutations have been described with continual new findings, demonstrating substantial genetic heterogeneity. Family screening is important when an index patient is identified; however, sporadic cases, as we see presented here, can also occur.
Clinical Characteristics in Common Genotypes of Congenital Long QT Syndrome
In an adolescent with seizure activity a normal EEG does not exclude a diagnosis of epilepsy, but the possibility of a nonepileptic cause of the convulsive episodes should be considered. Of note, clinical studies10,11 have shown that up to one-third of presumed “resistant epilepsy” may be variants of LQT syndrome. Given the increasing recognition of this syndrome and its risks, it should be included in the differential diagnosis of “resistant epilepsy” among young people. In this clinical situation, the ictal episodes can be characterized by continuous ECG monitoring. In case of frequent episodes, an ictal EEG/ECG-video recording can be a useful tool.12 With recognition and appropriate treatment of affected patients, the potentially fatal consequences of LQT syndrome may be prevented.
Notes
This article was externally peer reviewed.
Funding: none.
Conflict of interest: none declared.
- Received for publication May 23, 2008.
- Revision received September 14, 2008.
- Accepted for publication October 6, 2008.



{kind=link}
{kind=link}
{kind=link}