
Abstract
Background: Acetylcholinesterase inhibitors are the first drugs to alter the devastating effects of Alzheimer disease. The next generation of drugs will prevent the β-amyloid plaques and neurofibrillary tangles or block enzymes that lead to neuron destruction. Effective use of these medications will require early identification of patients at risk.
Methods: Using the PubMed service of the National Library of Medicine, all English language articles published in 2000, 2001 and the first half of 2002 with a key word of ‘dementia’ were reviewed for articles that described the emerging pathophysiologic model for Alzheimer disease.
Findings: Standardized clinical screening tools, such as the mini-mental status examination and the clock test, administered longitudinally and correlated with family observations, can identify many at-risk patients. Genetic testing can identify a known mutation in 70% of patients who have a high family incidence of Alzheimer disease but awaits effective prevention before being useful. The molecular mechanisms of Alzheimer disease will eventually lead to prevention.
Conclusion: Today, these patients benefit from nutritional support and lifestyle enhancement encouraged through a continuous primary care relationship.
Alzheimer disease is associated with an excessive number of extracellular β-amyloid plaques and intracellular neurofibrillary tangles in the cerebral cortex and subcortical gray matter. These plaques and tangles cause structural brain damage and extinguish cognitive functions in more than 3 million Americans and affect 16% of those older than 85 years.1–4 One in 10 elderly Americans has mild cognitive impairment and is at risk for Alzheimer disease.5 Today, acetylcholinesterase inhibitors temporarily delay decline for some patients, but effective interruption of progression remains elusive. Biomolecular advances over the last 2 years have uncovered the events that lead to the production of plaques and tangles, revealing opportunities for intervention. Future pharmaceuticals will depend on understanding the pathogenic and molecular cascade leading to Alzheimer disease.
We will review the emerging molecular theories, genetics, early diagnostic techniques, and promising interventions of Alzheimer disease and correlate the new science to primary care. The goal is to facilitate the family physician when addressing questions posed by patients and families seeking solace for the alarming diagnosis of Alzheimer disease.
Methods
Using the PubMed service of the National Library of Medicine, all English language articles published in 2000, 2001, and the first half of 2002 with a key word of ‘dementia’ were identified. Articles describing advances in the understanding of the molecular basis, treatment, screening, and genetics of Alzheimer disease were further reviewed. Those findings that were confirmed in more than 1 research publication by different researchers or referenced in 2 review articles appearing in peer-reviewed journals were selected for further analysis to construct a “prevailing” theory of the molecular pathogenesis of Alzheimer disease. The most recent or the most descriptive article has been referenced. Salient literature published before 2000 was accessed if referenced by current literature, in an attempt to focus on the theorems that remained current. The level of evidence for clinical recommendations were judged by the authors as follows: level A is supported by randomized clinical trial or meta-analysis, level B by other evidence, and level C by consensus or expert opinion.
Results
The Emerging Molecular Theory
Four major processes contribute to the brain destruction presenting clinically as Alzheimer disease. They are amyloid production, tangles formation, inflammation, and neurodegeneration or cell death.6 Each process contributes to and interrelates with the others in a maze of cause and effect that is still being elucidated.
Amyloid
Two decades of research have confirmed the significance of plaques formed from β-amyloid deposits in the extracellular spaces of the brain. These destructive plaques act like barnacles attaching to neurons. They are formed from a injurious mix of β-amyloid, inflammatory cells, and free radicals.7 The brains of aged, cognitively normal humans often contain a diffuse pattern of amyloid deposits, but few plaques. In Alzheimer disease, the plaques accumulate in the critical parietal, temporal cortex, and hippocampus areas associated with memory and learning functions. Much like fatty streaks organize into atherosclerotic plaques that damage arterial walls, amyloid deposits in some patients can organize into β-amyloid plaques that damage the cell walls of neurons.8
β-Amyloid is generated from large structural molecules called amyloid precursor proteins.9 Amyloid precursor proteins are transmembrane glycoproteins that play a role in cellular adhesion and membrane integrity. They are found in intracellular vesicles, such as the endoplasmic reticulum, Golgi apparatus, endosomes, and neurotransmitter secretory tubules, as well as the cell wall. Enzymes (secretases) on membrane surfaces cleave amyloid precursor proteins to form amyloid and other protein fragments that promote membrane function and also signal DNA transcription activity in the nucleus. Amyloid is present in the cerebrospinal fluid of healthy humans (and mice), but greater quantities are found in patients with Alzheimer disease.
Some secretases on membrane surfaces, using a 2-step process, cleave these amyloid precursor protein fragments into the insoluble β-amyloid.8 β-Amyloid formed in the cytoplasm of the cell is taken up by vesicles that fuse with the cell membrane and release into the extracellular space. Here, in Alzheimer disease, they aggregate into plaques.
β-Amyloid plaques activate astrocytes and glial cells, releasing cytokines, and acute phase reactants, and they trigger an inflammatory cascade. Binding of complement triggers altered ionic homeostasis, excessive calcium entry into neurons, hyperphosphorylation of τ-protein, neuronal dysfunction, loss of synapses, pruning of dendrites, decreased neurotransmitter release (especially acetylcholine), and cell death. All the downstream effects of β-amyloid plaque accumulation have been difficult to place into their proper temporal sequence.8 However, the load and distribution of plaques formed from β-amyloid correlate directly with the severity and manifestation of Alzheimer disease.
Neurofibrillary Tangles
Neurofibrillary tangles are also pathognomonic for Alzheimer disease. They occur in large numbers within the cytoplasm of neurons, particularly in the frontal, temporal, and parietal cortex, hippocampus, and the amygdala.8 Tangles are paired helical filaments made of the remnants of damaged intracellular microtubules and hyperphosphorylated τ-protein. τ-Protein is a multifunctional protein that plays a role in the assembly and stabilization of microtubules. When τ-protein is hyperphosphorylated, the tubules twist into compact filamentous structures and act like foreign bodies within the cell (Figure 1). Two enzymes, both kinases (Cdk5 and Csk-3β) are overly active in Alzheimer disease, with β-amyloid playing a role in their activation.10

Secretase and Alzheimer disease. Amyloid precursor proteins (APP) serve active and passive structural functions in the cell wall of neurons. To be activated, they are cleaved by α-secretase fragments that may play a role in cell adhesion. β-Secretase cleaves APP at a different site, producing a 42-amino acid fragment that can further be acted on by γ-secretase to produce β-amyloid, a protein fragment with low solubility that can aggregate and adhere to the neuron cell wall causing damage.
Protease Enzymes and Amyloid
The enzymes that fragment the amyloid precursor proteins are called secretases. Precursor proteins are split in sequence. The first sequence can be at 1 of 2 sites, by 2 different secretases (α- or β-secretase). Both secretases are found in most membranes, including the Golgi apparatus, endosomes, and the plasma membrane. Only β-secretase, also called BACE, creates the substrate from which β-amyloid can be produced. It is unclear whether both secretases are essential, because mice bred to be devoid of β-secretase seem normal except that they do not accumulate β-amyloid.11
As shown in Figure 2, there is a further cleavage necessary to produce β-amyloid. γ-Secretase further splits the fragments of amyloid precursor protein produced by β- and α-secretase. When the fragments created by β-secretase are acted on by γ-secretase, the insoluble β-amyloid, a 42-amino acid fragment, is produced. Presenilins are cofactors of γ-secretase or, as recent evidence suggests, they actually are γ-secretase. Altered production of presenilins, as seen in some at-risk families, results in an increase of β-amyloid.11

τ-Protein phosphorylation. The role of τ-protein in the cell is to provide structural support to microtubules. In the presence of kinase, τ can become phosphorylated, causing microtubules to disorganize and form tangles that disrupt intracellular transport. Phosphorylation seems to be influenced by the presence of β-amyloid.
Pharmaceutical researchers are actively pursuing drugs to block β- or γ-secretase or enhance α-secretase.9 There are also several cofactors necessary to activate secretase that may be manipulated to decrease the formation of β-amyloid. These include the cell surface immunoglobulin molecule receptor for glycation end products (RAGE) and β-amyloid binding alcohol dehydrogenase (ABAD).12
Inhibiting the phosphorylation of τ-protein by kinase enzymes is another potential target for prevention. The kinase Cdk5 plays a critical role in brain development, neurogenesis, dopamine signaling, and synaptic vesicle release, but selective inhibitors of Cdk5 and GSK3β, such as lithium, have demonstrated limited protection against neuronal death in cell culture.10 However, because of its essential role in brain development, enthusiasm for kinase inhibition has been restrained. It is also generally accepted that τ phosphorylation is a downstream event to β-amyloid production.6
Microischemia and Amyloid
Dementia, including Alzheimer disease, is more likely to develop in persons with cerebrovascular disease.13,14 Ischemia in the microcirculation of the temporal lobes of patients with Alzheimer disease results in low oxygen tension and β-amyloid production, much as ischemia causes amyloid deposits in the heart.15 Supporters of ischemia theories of Alzheimer disease postulate that amyloid precursor protein production increases to protect the cell membranes from the oxidative stress of ischemia.16 More amyloid precursor protein begets more β-amyloid, causing the clinical abnormalities seen in Alzheimer disease.16
Apolipoprotein and Amyloid
Apolipoprotein E plays a role in the transport of cholesterol and phospholipids. It is encoded by a gene on the long arm of chromosome 19. There are 3 isoforms, each of which codes for a slight variation in the amino acid sequence. The E4 allele increases the susceptibility to Alzheimer disease through increased disposition of amyloid plaques and decreased clearance of β-amyloid. In the brain, apolipoproteins are essential for cytoskeletal integrity and neuronal repair mechanisms, making them unfavorable targets for manipulations to alter Alzheimer disease.17,18
Homocysteine is associated with cardiovascular disease and stroke and is emerging as a risk factor for Alzheimer disease. It promotes amyloid-peptide-mediated toxic effects in neuronal cell cultures and induces apoptosis in rat neurons. In a prospective study of 1000 nondemented Framingham residents over the age of 76 years, not only were homocysteine levels predictive of Alzheimer dementia but also the higher the level, the greater the risk.19 The impact of homocysteine was not altered by educational levels, systolic blood pressure, smoking, alcohol intake, diabetes, obesity, stroke, or the apolipoprotein E genotype.19
Amyloid and Neurotransmitters
Degeneration of the neurons and loss of dendrites profoundly reduces the production of acetylcholine. A loss of 60% to 90% of acetylcholine activity results in memory impairment. Other neurotransmitters depleted in Alzheimer disease include serotonin by 50% to 70%, somatostatin by 40% to 60% and norepinehrine by 30% to 70%. Receptors for serotonin, glutamate, somatostatin, and acetylcholine are reduced also.20 Symptomatic treatment of Alzheimer disease has focused on augmenting neurotransmission by increasing the brain’s cholinergic supply. Greatest success has been obtained by blocking the enzyme acetylcholinesterase, which clears acetylcholine from the synapse. Tacrine, donepezil, rivastigmine, and galanthamine, all of which are acetylcholinesterase inhibitors, have confirmed efficacy (level A) in temporarily delaying the progression of Alzheimer dementia.21
Inflammation
All pathologic studies in Alzheimer disease confirm the accumulation of microglia around β-amyloid plaques.22 Microglial cells clean up debris and cellular damage in the brain but in the process release free radicals and cytokines that activate astrocytes. Astrocytes, the largest cell population in the brain, provide metabolic support to neurons but add to the oxidative imbalance. Consequently, an inflammatory cascade is initiated.10
Numerous anti-inflammatory approaches have been studied. Prednisone has not been effective in Alzheimer disease, and celecoxib, the selective cyclo-oxygenase inhibitor, has not been shown to arrest disease progression but is epidemiologically associated, as are other nonsteroidal anti-inflammatory drugs (NSAIDs), with a protective effect.6
Genetics
Only 10% to 20% of cases suggest a familial mode of transmission, but understanding phenotypes produced by these mutations has contributed a great deal to the understanding of Alzheimer disease. Mutations in 4 genes have been affirmed to increase risk for Alzheimer disease, but other mutations are in various stages of study.23 First, the amyloid precursor protein gene, found on chromosome 21, results in increased production of precursor proteins and may explain the prevalence of Alzheimer disease in Down syndrome.11
Second, the Presenilin 1 gene, located on chromosome 14, increases the activity of secretase and accounts for 4% of early-onset Alzheimer disease cases between the ages of 28 and 50 years. The third mutation, Presenilin 2, on chromosome 1, accounts for 1% of Alzheimer disease cases with onset between 40 and 50 years of age.24 The presenilin mutations increase levels of γ-secretase (and in fact may be γ-secretase) and result in an aggressive form of inherited Alzheimer disease.25
The fourth and most common form of inherited Alzheimer disease is associated with one of several mutations on chromosome 19, the apolipoprotein E alleles. These mutations cause enhanced aggregation and decreased clearance of amyloid peptides in the brain. Their expression is variable, but they account for 50% of early Alzheimer disease and 20% of late-onset disease. The presence of the ϵ 4 allele mutation increases the risk for Alzheimer disease in whites and ϵ 2 and 4 alleles increase the risk in African Americans. Apolipoprotein genotyping is commercially available but offers little advantage as a prognostic indicator. Female carriers have a 45% probability of developing Alzheimer disease by age 73 and male carriers have only a 25% probability. Genetic testing is useful only with atypical presentations of Alzheimer disease.26,27 The heterogeneity in late-onset, sporadic Alzheimer disease complicates interpretation of genetic profiling.28
Currently, a DNA diagnosis is possible in 70% of families with a pattern of autosomal-dominant Alzheimer disease. For first-degree relatives of carriers, lifetime risk (onset by age 90) is increased some 3- or 4-fold relative to control subjects.29 Patient response to genetic testing is not well studied, but with proper counseling, most patients demonstrate effective coping skills and find testing constructive.30 Testing concerns include expense, accuracy, limited understanding of risk calculations, insurance denials, emotional impact, and deterministic behavior leading to unnecessary risk exposure. There is very little clinical evidence to support genetic testing for a known Alzheimer disease mutation and no evidence to support the use of genetic testing as a screening tool.
Early Diagnosis
Identifying patients at risk for Alzheimer disease, and developing strategies that modify the disease are the most urgent challenges facing researchers. Disease modification is likely to be of greatest benefit if begun early. Defining clinical patterns and searching for molecular markers of preclinical Alzheimer disease are active areas of research.
Clinical Identification of Early or pre-Alzheimer Disease
Clinical risk factors for Alzheimer disease include advanced age, malnutrition, small head size, head trauma, and female sex. Decreased sensory responsiveness caused by declines in vision and hearing can lead to decreased awareness, and depression, putting the patient at risk for dementia. Self-reported poor general health is associated with a 5-fold increase in the development of Alzheimer dementia.31 Many elderly patients have mild cognitive impairment but use compensatory strategies in everyday living. Only 20% of these patients will progress to dementia.32
Motor skills decline can also be predictive. Subjects who later develop cognitive impairment have slower finger tapping, take longer to walk 30 feet, and have an inability to suppress the eye-blink response to sudden movement.33–35
Framingham Study participants have confirmed difficulty with verbal memory, a temporal lobe activity, as an early sign of Alzheimer disease.36–39 Families perceive verbal memory problems as forgotten telephone arrangements but compensate by suggesting the date be written down. On the Mini-Mental Status Examination, recalling 3 objects is verbal memory-sensitive.40
Clock Test
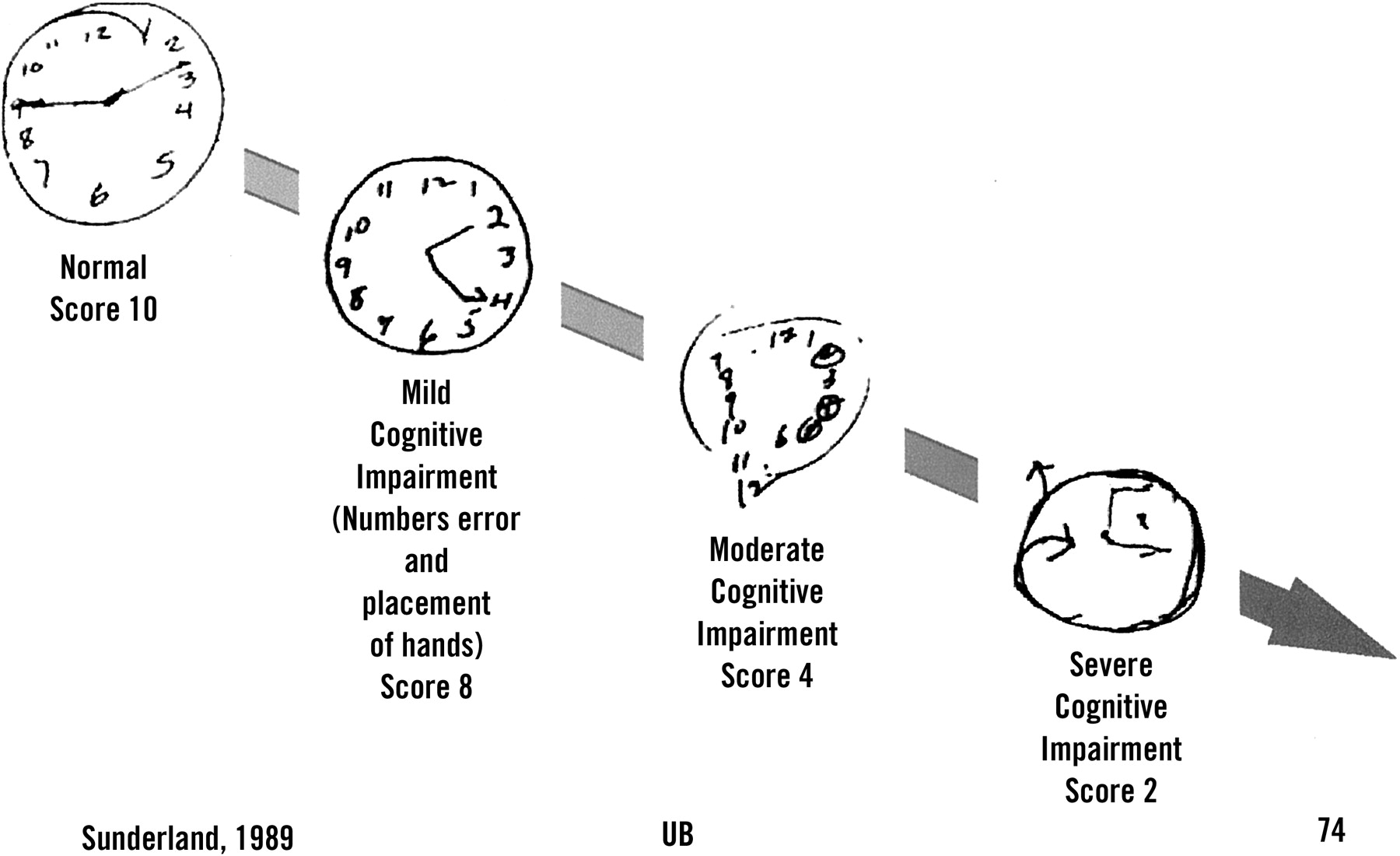
The search for a standardized scoring system capable of reliably predicting dementia remains elusive.41,42 The clock-drawing test is a simple measure of visuospatial ability; there is relatively little overlap between patients with Alzheimer disease and healthy control subjects early in the disease (Figure 3).43 It is quick and easy to administer. Recent studies suggest the clock test provides 85% sensitivity and 85% specificity and correlates well with other cognitive tests (level B).44

Clock drawing test. The patient is asked to draw clock displaying the time 2:45. This simple test has been shown to be more sensitive of early Alzheimer dementia than several other screening tools and can be scored according to standard protocols.43
Unfortunately, the use of cognitive scales in an unselected elderly population produces many false positives because of significant cultural, mood, or education variation.45–47 Standardized scales are most useful for evaluation of patient or family concerns about cognitive decline. If the clinician cannot substantiate a deficit during clinical testing, progressive deterioration is much less likely.48
There is level B evidence to recommend that a patient who is identified to be at risk for Alzheimer disease should undergo cognitive measurement by a standardized scoring system such as the clock test or the Folstein Mini-Mental Status Examination. This scoring should be repeated every 6 to 12 months thereafter. If there is a decline of 3 points, the patient should be considered at risk for the onset of dementia and evaluated for treatment.49
Laboratory Identification of pre-Alzheimer Disease
The goal of a laboratory work-up in a patient with suspected Alzheimer disease is to assure an accurate diagnosis of the disorder responsible for the dementia and discover correctable conditions. The treatment plan is altered in up to 13% of patients after a laboratory work-up.50,51 Patients should have a complete blood count, electrolyte panel, a comprehensive metabolic panel, thyroid function tests, syphilis serology, vitamin B12 and folate levels, and pre-albumen (level B).52,53 Determination of erythrocyte sedimentation rate, HIV antibody screen, toxicology screen, and chest radiograph should be ordered if indicated by history or physical examination.52
Advertisements for a urine screen capable of diagnosing Alzheimer disease have appeared in the lay press. This screen tests for breakdown products of arachidonic acid that appear in the urine of patients with Alzheimer dementia.54 Overlap between healthy elderly patients and those with Alzheimer dementia renders this test too nonspecific for clinical recommendation.
Several substances in blood and cerebrospinal fluid are under study as diagnostic markers for Alzheimer disease.6 Autoantibodies to β-amyloid and neurotransmitters have been demonstrated in the blood of patients with Alzheimer disease.55,56 Melanotransferrin (p97) originates from the microglia activated by the β-amyloid plaques and can be detected in the cerebrospinal fluid.57 There are elevated levels of β-amyloid and τ-protein in cerebrospinal fluid and high levels of β-amyloid in plasma. None of these markers are yet available for clinical use.58,59
Imaging in pre-Alzheimer Disease
Most patients should undergo computed tomography (CT) or magnetic resonance imaging (MRI) of the head.52,60 Not only are other conditions ruled out by imaging but also decreased hippocampal volume precedes Alzheimer disease in 50% to 70% of patients.61 The association between brain volume and function remains significant after adjusting for age and has been confirmed in African Americans and white persons.62–66 Positron emission tomography (PET) to detect accumulation of β-amyloid plaques in memory-related brain regions correlate with disability and are 93% sensitive and 76% specific.67,68
As we have shown, routine screening with available tools is not cost-effective.69 Clinical suspicion aroused during continuity care confirmed by focused clinical and laboratory testing remains the best way to identify patients at risk for Alzheimer disease.70
Altering the Course of Alzheimer Disease
Maintaining the general health status of elderly patients is the most effective strategy to lessen the impact of Alzheimer disease on a population. Table 1 lists strategies that have been proposed along with available evidence. Modifying cardiovascular risk factors, such as hypertension and high cholesterol, and homocysteine levels impact both heart disease and dementia (level B).19,71 Continued intellectual and occupational achievement, even memory games, increase dendrites and delay clinical manifestations.72 Good nutrition enhances functional speed and brain efficiency.72
Strategies to Slow the Onset or Progression of Dementia
Cholinesterase inhibitors induce modest improvement in cognitive function for a limited span of time.73 Although indicated in mild to moderate disease, they are not neuroprotective and offer little clinical improvement when the Mini-Mental Status Examination score drops below 12.74 In the near future, neuroprotective approaches will focus on amyloid precursor protein, amyloid metabolism, and general neurodegenerative processes such as inflammation and oxidation.75
Decreasing β-Amyloid Accumulation
All humans experience a continuum of β-amyloid production and clearance, but in Alzheimer disease, plaques damage dendrites, stimulate the inflammatory response, and kill neurons. Tangle formation, universal to Alzheimer disease, is probably secondary to β-amyloid production. It is likely that in the next few years, drugs capable of inhibiting β- and γ-secretase will emerge.
The neuron membrane-bound enzyme β-secretase is a prime target for drug development because of its critical instigating role in the formation of β-amyloid (Figure 2). It was identified in 1999 and appears in the literature under several names (ie, BACE1, Asp2, memapsin2). Mice that lack β-secretase do not produce brain β-amyloid but develop normally, increasing the optimism that suppressing β-secretase activity has potential. Several nonpeptide, small-molecule inhibitors of β-secretase capable of crossing the blood-brain barrier are under development.6,75
Genetic mutations (PS1, PS2) result in altered structure of γ-secretase and result in early-onset Alzheimer disease. γ-Secretase cleaves amyloid precursor proteins in cellular and subcellular membranes (Figure 2). However, γ-secretase “knockout” mice die in utero, raising caution among researchers. Still, several modulators of γ-secretase activity, such as nicastrin, selectively inhibit amyloid precursor breakdown and decrease β-amyloid production.6,75,76
Immunotherapy
Immunotherapy has been met with enthusiasm until recently. Antibodies to β-amyloid increase its clearance from the brain. In mice, prophylactic immunization, even by a nasal application of a β-amyloid fragment, reduces β-amyloid plaque and tangle formation and protects against learning and memory impairments.77–79 Unfortunately, a β-amyloid immunization trial in humans had to be discontinued because of a high incidence of encephalitis in treatment subjects. Human subjects selected for these preliminary studies already had Alzheimer disease with a significant load of β-amyloid before enrollment. It is possible that the antibody-antigen reaction induced resulted in the encephalitis-like event. More studies will follow.
Inflammation
Vitamin E (α-tocopherol), a lipid-soluble vitamin antioxidant, traps free radicals like those present in β-amyloid plaques. In animal studies, vitamin E reduced degeneration of hippocampal neurons; in humans, it has been demonstrated to slow Alzheimer disease progression, although it did not improve function. The dose ranges studied were between 400 IU and 1000 IU twice per day (level A).80,81
Other antioxidants, such as ginkgo biloba, vitamin C, selegiline (an monoamine oxidase inhibitor with an antioxidant effect), and nicergoline have shown inconsistent results that range from no benefit to minimal delayed progression of Alzheimer disease.80,82,83
Estrogen has a weak antioxidant effect and has been shown to alter cholinergic, serotonergic, and catecholaminergic neurotransmitter systems. Men convert testosterone to estrogen in the brain and are less likely to experience late-life estrogen deficiency. Several short-term estrogen supplemental studies in women demonstrated improved mood and psychologic function over 6 to 12 weeks, but others failed to show any effect after 12 months.84,85 The Women’s Health initiative, a prospective study, has identified a slightly increased risk of dementia in the estrogen/progestin group, but this may be due to increased atherosclerotic vascular disease.86 One published meta-analysis suggests there is possibly a cognitive benefit to estrogen but recommends the use of estrogen only in women with other indications for hormones supplemention.87 Another more recent longitudinal study demonstrated a clinically significant reduction in dementia for both men and women taking estrogen (inconsistent).88
NSAIDs have been associated with lower risk and slower rate of decline in Alzheimer disease but only if they are started in the preclinical phase and used for a minimum of 2 years (level B).22,89 The largest studies to date have been observational. The varying dosages used in these studies makes it difficult to confidently recommend a minimum effective dose, but a sub-anti-inflammatory dose seem to be protective for some agents.90 Acetaminophen and prednisone have no effect, and aspirin has less effect than NSAIDs.20 One 12-month trial failed to show a protective effect of celecoxib.6
Treating Cholesterol
People with systolic hypertension, high low-density lipoprotein (LDL), or high apolipoprotein E in midlife have 3 times the risk of Alzheimer disease in later life.14,91 Epidemiologic studies have demonstrated that 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) used to control hyperlipidemia are associated with a 60% lower incidence of Alzheimer disease.92,93 There is mounting evidence that lowering serum cholesterol retards the onset of Alzheimer disease.94 The role of folic acid in the reduction of homocysteine is also interesting.
For inflammation and hypercholesterolemia, the observations are mostly epidemiologic. Although ongoing intervention trials will further define treatment effect, there is ample evidence that aggressive treatment of hypertension and hyperlipidemia prevents atherosclerosis (level B). There is mounting evidence that antioxidants and anti-inflammatory agents can alter disease onset (level B).95 It is likely that success in management of chronic diseases will protect some patients from Alzheimer disease.81
Conclusions
Alzheimer-type dementia remains a clinical diagnosis. However, neuroimaging by CT or MRI with attention to the hippocampal region in the initial evaluation of patients with dementia is appropriate for confirmation and staging. Currently, no genetic markers can be recommended for routine diagnostic purposes; however, screening for depression, B12 deficiency, and hypothyroidism should be performed on most patients.52 Treatment should include multiple modalities, just as prevention of atherosclerosis does now. Active lifestyle and vitamin E can be recommended for most elderly patients. NSAIDs and estrogen may be recommended to patients with coexisting indications. Acetylcholinesterase inhibitors should be discussed as a palliative treatment when decline is detected on a standardized score. The family physician should be optimistic that effective preventative modalities will be released in the next several years. Effective use of newer agents will be dependent on the family physician’s first-hand knowledge of the patient’s baseline.
- Received for publication December 13, 2002.
- Revision received December 13, 2002.
References



{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.