
Abstract
Background: Acute pancreatitis as the initial manifestation of systemic lupus erythematosus (SLE) has been documented only nine times in the English literature.
Methods: We report the case of a 25-year-old woman patient with fever, abdominal pain and vomiting, elevated levels of pancreatic enzymes, and various other laboratory abnormalities. Further investigation led to a diagnosis of SLE. A literature search, using the key words “systemic lupus erythematosus” and “pancreatitis” was undertaken.
Results and Conclusions: The treatment of SLE pancreatitis is steroids, which is somewhat controversial because steroids have been implicated in the cause. SLE can involve any organ system. It is important that the family physician, who treats patients as a whole, rules out SLE when a straightforward diagnosis is associated with inexplicable multiple concomitant abnormalities.
Systemic lupus erythematosus is a multisystemic autoimmune disorder characterized by a broad range of manifestations and the finding of antibodies in the blood directed against one or more components of cell nuclei.1 It has a definite female predominance, especially after the onset of puberty, but has been known to occur in children as young as 3 years of age.2 The most common areas of involvement are the joint and cutaneous system, with nonspecific complaints of fever, malaise and fatigue, and renal disease.3 The initial manifestation, however, can involve any organ system either singly or in combination, which frequently makes diagnosis difficult. The American Rheumatism Association recommends 4 of the following 11 revised criteria for the diagnosis of SLE: malar rash, discoid rash, photosensitivity, oral ulcers, arthritis, serositis, renal disorder, neurologic disorder, hematologic disorder, immunologic disorder on serologic testing, and antinuclear antibodies.4
Methods
A case of a 25-year-old woman patient with fever, abdominal pain and vomiting, elevated levels of pancreatic enzymes, and various other laboratory abnormalities is reported. Further investigation led to a diagnosis of SLE with pancreatitis as its initial manifestation. A literature search using the key words “systemic lupus erythematosus” and “pancreatitis” was undertaken.
Case Report
A 25-year-old African American woman, previously well, came to the University Hospital of Brooklyn complaining of a 2-week history of lower abdominal pain and occasional dysuria. She also related a 1-week history of fever, loss of appetite, generalized weakness, a nonspecific cough, and a single episode of vomiting on the day of her visit. She had been to 2 different physicians within the previous week, who had prescribed various antibiotics for a presumed diagnosis of upper respiratory tract infection and pelvic inflammatory disease, respectively.
Her medical history was notable for childhood illnesses of atopic dermatitis, migraines, and occasional nonspecific chest pains. She also received treatment for gonorrhea and chlamydial infection when she was 20 years old. She denied alcohol use and illicit drug use. Her family history was unremarkable for autoimmune disorders. She had started oral contraceptives 3 months earlier.
She reported some thinning of her hair for the past 4 months, a weight loss of 27 pounds within the past 3 months, and bilateral thigh pain and shoulder pain for the past 2 weeks that resolved in the few days before her visit. The patient attributed her recent weight loss and leg pain to an intensive physical training program in which she was enrolled during the past 3 months.
When examined, she was a young, alert, well-appearing woman. She was afebrile, her pulse rate was 100 beats per minute, and her blood pressure was 104/64 mmHg. She had no skin lesions or lymphadenopathy. Her eyes were normal on examination. Her chest sounds were clear with no friction rubs, and she had no murmurs or rubs. Her abdomen was soft with mild suprapubic tenderness, minimal guarding, and no rebound tenderness. Bowel sounds were present. She had no vaginal discharge, cervical motion tenderness, or adnexal masses. Her uterine size was normal. Her extremities showed no joint swelling or tenderness. Neurologically, she was unremarkable.
Laboratory studies disclosed the following values: sodium 139 mEq/L, potassium 3.8 mEq/L, chloride 105 mEq/L, bicarbonate 21 mEq/L, blood urea nitrogen 10 mg/dL, creatinine 0.6 mg/dL, glucose 84 mg/dL, aspartate aminotransferase 119 U/L (normal 14–50 U/L), alanine aminotransferase 76 U/L (normal 10–65 U/L), protein 8.2 g/dL, albumin 3.7 g/dL, calcium 8.5 mg/dL, and total bilirubin 1.3 mg/dL (normal 0.2–1.3 mg/L). A complete blood count showed a white cell count of 1.62 ×103/μL with a differential cell count of neutrophils 73%, band cells 15%, lymphocytes 15%, monocytes 5%, and atypical lymphocytes 2%. Hemoglobin was 11.8 g/dL, hematocrit 35.8%, and platelets 183,000/μL. Pancreatic enzymes determinations disclosed an amylase level of 134 U/L (normal 30–110 U/L) and a lipase level of 712 U/L (normal 23–300 U/L). Urinalysis showed cloudy urine with protein 300 mg/dL, a white cell count of 12–20 per high-power field (HPF), granular casts 1–2/HPF, bacteria loaded, and many epithelial cells. Nitrite and leukocyte esterase were negative. Urine pregnancy test was negative.
A computed tomographic (CT) scan of the abdomen with contrast showed no evidence of gallbladder stones or abnormality of the pancreas. A sonogram on day 3 of admission showed a possible left pleural effusion, but otherwise findings were normal.
The patient was admitted to the hospital with a differential diagnoses of urinary tract infection, neutropenia secondary to human immunodeficiency virus (HIV) infection or acquired immunodeficiency syndrome (AIDS), and possible perihepatitis complicating pelvic inflammatory disease. She was allowed nothing by mouth, given intravenous fluids, and started empirically on antibiotics. By day 3 of admission her serum lipase had doubled to 1445 U/L and her white cell count had dropped to 0.92 × 103/μL. Her gastrointestinal discomfort had resolved, and she reported feeling better.
Connective tissue workup showed a antinuclear antibody (ANA) titer 1:1280, anti-double-stranded DNA antibody positive, anti-DNA titer 1:640, anti-Smith antibody positive, anti-ribonucleoprotein positive, anti-SSA and anti-SSB negative, complements C4 < 10 mg/dL (normal 16–47 mg/dL), C3 16 mg/dL (normal 83–201 mg/dL), anticardiolipin antibodies were elevated, immunoglobulin G (IgG) was 23 g/L (normal 0–14 g/L) and IgM 24 mg/L (normal 0–9 mg/L). Lupus anticoagulant was negative. Tests for gonorrhea, chlamydial infection, and HIV were all negative. A diagnosis of SLE pancreatitis was made on the fourth day of admission, and steroids were started. Pancreatic enzyme levels continued to rise, peaking on day 12 of admission despite continuous steroid therapy and strict orders of nothing by mouth.
The response of her pancreatic enzymes to steroids is shown in Figure 1. The patient’s temperatures spiked to 104.8 °F throughout her 34-day hospital stay despite negative urine, blood, and sputum cultures. Her elevated temperatures continued despite withholding her antibiotics for possible drug fever. Repeated CT scans were performed on days 13, 25, and 33 to rule out microabscesses as a source of her fever, but no pancreatic pathologic lesions were seen.
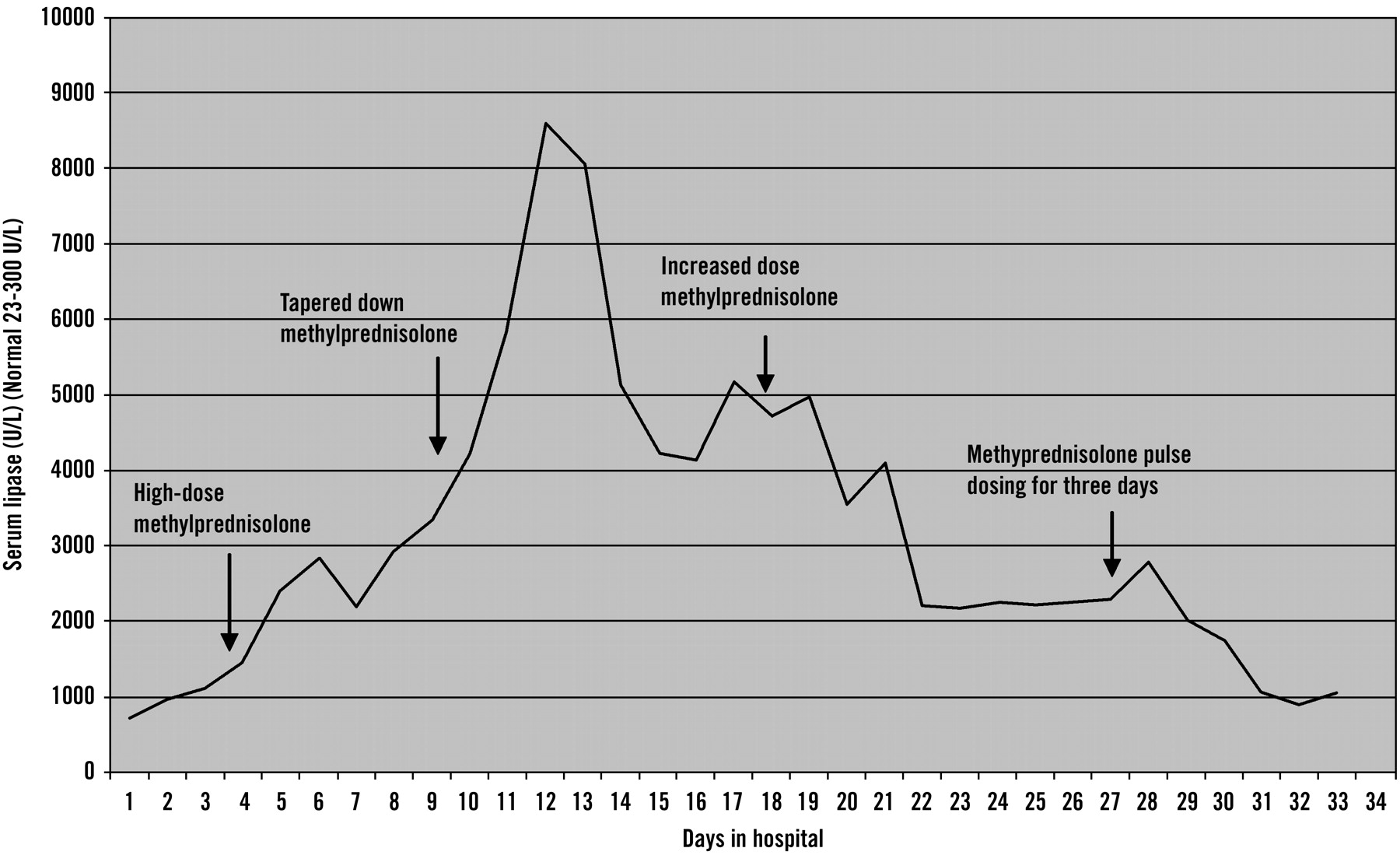
Graph depicting the response of serum lipase levels to differing doses of methylprednisolone therapy.
The patient’s leukopenia responded only transiently to filgrastin, a granulocyte colony-stimulating factor, given on three occasions. During her steroid therapy she developed a discoid rash in both ears and an oral ulcer, both of which disappeared with continued steroid therapy. She was discharged after near normalization of her pancreatic enzymes (amylase 73 U/L, lipase 657 U/L) and disappearance of the temperature spikes. She received a tapering dose of oral prednisone on discharge.
Her discharge diagnosis was SLE pancreatitis. When she returned for a follow-up visit after 3 months, she had normal pancreatic enzymes (amylase 88 U/L, lipase 291 U/L), with corresponding normalization of complement (C3 100 mg/dL, C4 30 mg/dL). Her white cell count was normal at 5.27 × 103/μL, and a urinalysis was negative for protein and casts.
Discussion
Our case illustrates the protean manifestations of SLE and a rare initial manifestation as acute pancreatitis. In retrospect, although our patient fulfilled the ARA criteria for the diagnosis of lupus on admission, the initial impression was an infectious process with HIV, given her young age, unmarried status, the history of sexually transmitted diseases, fever, and the degree of leukopenia. Fortunately, fortuitous screening with ANA titers for connective tissue disease by an ophthalmologist who had noted papilledema on funduscopy after she complained of eye floaters a few weeks earlier made for a timely diagnosis of SLE.
Our patient had multiorgan involvement of SLE, namely, hematologic, renal, and gastrointestinal, as well as involvement of the teguments; however, the most distinguishing feature of our patient’s condition was pancreatitis. Pancreatitis is an extremely uncommon manifestation of SLE. The association between SLE and pancreatitis was first documented in 1939 by Reifenstein et al.5 Since then, only 69 cases of SLE pancreatitis have been documented in the English literature, and only 9 of these cases, including ours, have had pancreatitis as an initial manifestation. 6,7 A diagnosis of SLE pancreatitis can be made only after the exclusion of other causes of acute pancreatitis, such as alcoholism and gallstones, the two most common causes in the United States. Viral causes such as in HIV and AIDS are considered in the immunocompromised patient.9
Since the initial description of SLE pancreatitis, there has been debate as to its origin, namely, steroid vs SLE as the primary cause. Most cases of pancreatitis in lupus have been in patients with long-standing SLE who have multiorgan involvement and are already on steroids, diuretics, or immunosuppressive therapy, all of which have been implicated in the etiology.10 The arguments against steroid-induced pancreatitis in SLE include the rarity of the occurrence of pancreatitis in non-SLE patients on steroids, the increasing documentation of pancreatitis as an initial manifestation of SLE, the finding of vasculitic lesions in the pancreas on autopsy, and the resolution of acute pancreatitis on continued steroid therapy.11,12 Recent studies support the role of SLE as the primary etiologic factor of SLE pancreatitis, with drug toxicity having some role.9,10 The mechanisms involved in the origin of SLE pancreatitis is believed to be vasculitis, microthrombi, and intimal thickening.10,12 Hypothetical mechanisms include complement activation, hypotension, and autoimmune reactions secondary to antipancreatic antibodies among others.13 It can occur both during a generalized flare and during disease quiescence, though the latter seems to be the tendency.12
The diagnosis of pancreatitis by itself is usually based on clinical findings of abdominal pain, nausea, and vomiting supported by laboratory findings of abnormal pancreatic enzymes and suggestive tomographic findings. Clinically the disease can manifest in any manner from a benign self-limiting disease to one with a fulminant course.
Within the context of SLE, besides the clinically evident cases of acute pancreatitis, there have also been cases of what is known as subclinical pancreatitis in which there is an elevation of pancreatic enzymes without clinical symptoms. The incidence of subclinical pancreatitis is believed to be much higher than clinical pancreatitis. One study found hyperamylasemia in 30.5% of asymptomatic SLE patients, suggesting that subclinical pancreatic damage might occur frequently in SLE.14
CT findings in acute pancreatitis, which usually correlate with the severity of disease, have a diagnostic accuracy of only 70% to 90%, even in severe pancreatitis.15 Our patient had repeatedly negative CT scans despite remarkably elevated pancreatic enzymes. As such, our diagnosis of acute pancreatitis was based primarily on the marked pancreatic enzyme elevation associated with the vague abdominal symptoms.
The treatment of SLE pancreatitis is with steroids. Steroid treatment is somewhat controversial because steroids have been implicated as the cause of SLE pancreatitis.10 Recent studies, however, have refuted the role of steroids in the origin of SLE pancreatitis and actually recommend the administration of steroids during the acute episode.12 Our patient showed an initial worsening in response to steroids and then a slow decline in pancreatic enzymes with continued steroid therapy (Figure 1). It is unclear whether the pulse dosing started on day 31 of admission had a more beneficial overall effect than continuous therapy. Additionally, in view of the initial poor response, it is difficult to determine whether the patient improved because of the steroids or whether improvement was just the natural course of the disease.
Conclusion
SLE can involve any organ system. It is important that the family physician, who treats patients as a whole, suspect SLE when a straightforward diagnosis is associated with inexplicable multiple concomitant abnormalities.
- Received for publication October 11, 2002.

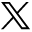


{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.