
Abstract
Introduction: Restless legs syndrome (RLS) is a common neurological disorder affecting 10% of the population. Most antidepressants exacerbate symptoms; however, correlational studies have noted symptom improvement with bupropion. The purpose of the current study was to examine whether, in a controlled study, bupropion would improve the symptoms of RLS, or at least not exacerbate them.
Methods: This was a double-blinded, randomized controlled trial. Twenty-nine participants with moderate to severe RLS received 150 mg sustained-release bupropion once daily, and 31 control participants received a placebo. Participants were followed for 6 weeks and completed standardized tools, including the International Restless Legs Syndrome Study Group (IRLSSG) severity scale.
Results: The primary outcome was change from baseline in IRLSSG severity score; lower scores were associated with improved symptoms. At 3 weeks, IRLSSG scores were 10.8 points lower in the bupropion group and 6.0 points lower in the placebo group (P = .016). At 6 weeks, IRLSSG scores were10.4 points lower in the bupropion group and 7.6 points lower in the placebo group (P = .108). Bupropion was more effective than placebo in the treatment of RLS at 3 weeks; however, this difference was not statistically significant at 6 weeks.
Conclusions: The data from our study suggest that bupropion does not exacerbate the symptoms of RLS and may be a reasonable choice if an antidepressant is needed in individuals with RLS. Larger studies that include titration of bupropion should be considered to determine if bupropion is appropriate for primary treatment of RLS, particularly considering the lower cost and favorable side effect profile compared with currently recommended first-line dopamine agonists.
Restless legs syndrome (RLS) is a common neurological movement disorder. It affects approximately 10% of the adult population of the United States and is more common with advancing age.1 RLS is diagnosed clinically based on 4 criteria (Table 1).2
Diagnostic Criteria for Restless Legs Syndrome
In addition to discomfort, individuals with RLS have overall worse quality of life than the general population,3 and depression and anxiety are more common among those with RLS.4 Sleep disturbances are also very common and are often the presenting complaint. Recent studies also show higher incidence of cardiovascular disease in patients with RLS.5
RLS may be a primary or a secondary condition. Secondary causes of RLS include iron deficiency, chronic kidney disease, pregnancy, and various medications. Treatment of the secondary causes may improve or resolve the symptoms of RLS.
Antidepressants, particularly serotonin-specific reuptake inhibitors (SSRIs), exacerbate RLS and periodic limb movement disorder, which is closely correlated with RLS. In one study, venlafaxine, citalopram, fluoxetine, paroxetine, and sertraline all caused increasing frequency of periodic limb movements, whereas bupropion decreased periodic limb movements compared with controls.6 In another study, RLS was noted as a possible side effect of the use of fluoxetine, paroxetine, citalopram, sertraline, escitalopram, venlafaxine, duloxetine, and mirtazapine.7 Finally, in one small case series, patients taking bupropion had improvement of their RLS symptoms.8 To date, no controlled clinical trials have studied the effect of bupropion on the symptoms of RLS.
Because depression is common in RLS and most commonly prescribed antidepressants exacerbate the symptoms of RLS, it would be beneficial to identify an antidepressant that did not exacerbate symptoms, particularly if it improved symptoms. If bupropion was found to improve, or at least not exacerbate, the symptoms of RLS, it would be a good choice for treating depression in individuals with RLS. We conducted a double-blind, randomized controlled trial comparing the effect of 150 mg sustained-release bupropion daily with placebo on the symptoms of RLS. We hypothesized that bupropion would improve the symptoms of RLS or at the least would not exacerbate RLS symptoms.
Methods
Overview of Study Design
The study was a randomized controlled trial of a bupropion regimen for adults with RLS. Active drug group participants were given a once-daily, 150-mg, sustained-release dose of bupropion for 6 weeks. Control group participants received identical placebo capsules. Outcome measures were assessed at baseline and weekly for 6 weeks.
Participants and Recruitment
Participant recruitment was accomplished via local radio advertisements, notices in local newspapers, and flyers placed in patient areas of academic medicine primary care clinics. To maximize recruitment, potential participants were screened for study eligibility using the RLS diagnosis questions presented in Table 1. Screening over the phone included positive responses to the 4 criteria in general terms. Criterion one (the urge to move legs usually accompanied or caused by uncomfortable or unpleasant sensations in the legs) was addressed over the phone with the question, Do you get an uncomfortable feeling in your legs associated with an urge to move? The form completed by the physician screener during the initial face-to-face evaluation listed each of the diagnostic criteria, with a choice of “Y” (yes) or “N” (no), which would be marked by the physician. For example, to determine whether criterion one was present, the screener would ask, “Do you have an urge to move your legs usually accompanied by uncomfortable or unpleasant sensations in the legs?” A potential participant had to answer affirmatively to all 4 questions and had to score 15 or higher on the International RLS Study Group (IRLSSG) scale to be eligible for the study (see below). In addition, participants had to be available for phone assessment weekly and be willing to travel to the study sites for in-person assessment at 3 and 6 weeks. Furthermore, potential participants had to be willing to accept randomization into either the active drug or placebo group.
Participants were excluded from study participation for the following reasons: history of seizure disorder, alcoholism, suicidal history or ideation, inability to return for 3- and 6-week assessment, no telephone access, eating disorders, age younger than 18, pregnancy, and unwillingness or inability to discontinue current medications for the treatment of RLS. Participants currently taking medications for the treatment of RLS were not automatically excluded from the study but had to complete a 2-week washout period off of the medication before becoming eligible. Information about exclusion criteria were obtained from potential participants by a study physician during a screening physical. The study physician was then responsible for determining participant eligibility for the study. The study was approved by the university institutional review board and informed consent was obtained consistent with federal guidelines. The study was registered with the Clinical Trials Registry before participant recruitment.
Procedures
After baseline data collection, eligible and consenting participants were placed in a pool and randomized in waves of 10 using a computer generated random list. Screeners were blinded to the allocation. Participants were randomized, in equal numbers within each block of 10, to either the active drug or control groups. Both groups received a bottle of 42 capsules with instructions to take one per day 2 hours before bedtime. The capsules given to the active drug group participants contained 150 mg sustained-release bupropion, and those given to the control group contained no active ingredients. Both groups received phone calls from study physicians at 1, 2, 4, and 5 weeks after baseline. Study physicians were blinded to randomizations. All participants also were scheduled to meet in the clinic with a study physician for more detailed assessment at 3 and 6 weeks after baseline assessment.
Measures
Basic demographic information was obtained from participants at baseline. Basic medical history was obtained as part of the screening physical. Restless leg severity was assessed using the IRLSSG scale. This validated 10-item questionnaire assesses symptom severity, frequency, and impact on daily life.9 The maximum score is 40, and a higher score indicates more severe RLS. Participants completed the IRLSSG during the in-clinic assessments at baseline, 3 weeks, and 6 weeks. The scale was also completed via phone at 1, 2, 4, and 5 weeks after baseline.
Depression was assessed via the Beck Depression Inventory (BDI-II). This 21-item scale is well-validated and commonly used to screen for depressive symptoms in both research and clinical settings. Scores can potentially range from 0 to 63, and higher scores indicate higher levels of depression. Study participants completed the BDI-II at baseline and at the 3- and 6-week assessments.
Finally, as part of every in-person and phone assessment after baseline, participants were asked how many days during the last week they had failed to take their study medication and whether they had experienced any problems with or had any concerns about the medication they were taking.
Data Analysis
Examination of potential attrition bias involved χ2 analysis and t tests using P < .05 as criteria for significance. Evaluation of baseline and study outcome differences by treatment group also used χ2 analysis and t tests where appropriate. All analyses of outcomes were intention to treat. Thus, all eligible participants who were initially randomized to a treatment group were included in all analyses, regardless of study completion status. For those lost to follow-up and missing a score at a particular time point, the most recent previous score for that participant on that measure was used in analysis.
Results
Study Completion
One hundred fifty-one adults were screened for the study during the period of January 2008 through February 2009. Seventy-two were determined to be eligible for the study, but 12 were excluded from the study and considered screening failures before obtaining any data from them. Reasons for considering participants as screening failures included no telephone or inability to contact at week 1 (n = 4), failure to complete washout (n = 2), inability to return to the clinic at 3 and 6 weeks (n = 3), or they chose not to participate before starting the medication (n = 3). The remaining 60 participants became the study sample, of which 29 were randomized to the active drug and 31 to the control group (see Figure 1).
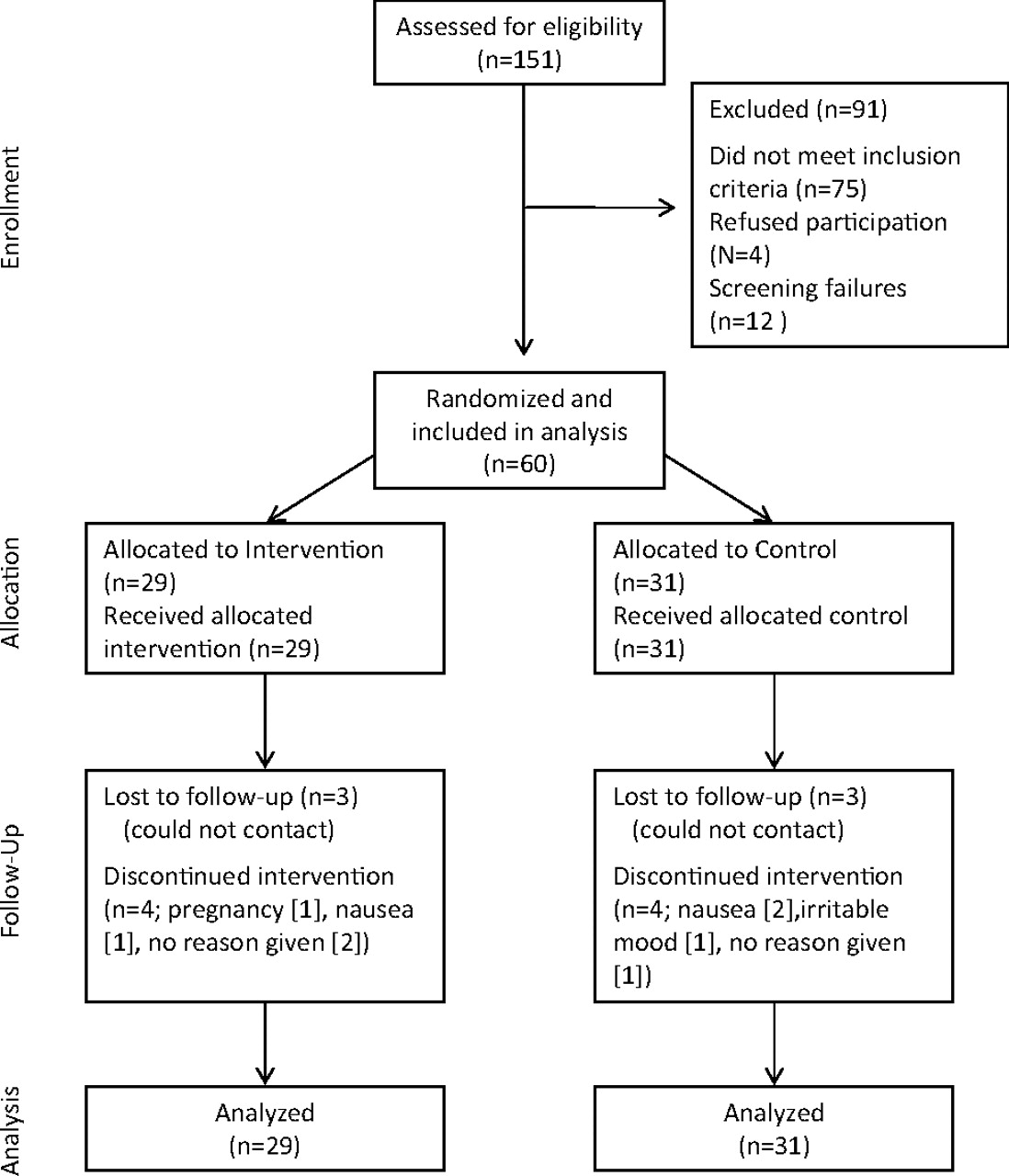
Participant flow diagram.
By the 3-week assessment, 11 of the 60 participants (18%) had either withdrawn from the study, could not be contacted, or failed to keep the 3-week clinic appointment. By week 6, an additional 3 participants were lost to follow-up, for a total participant completion rate of 77%. As shown in Table 2, those who failed to complete assessments at either time period did not differ significantly (P > .05) from those still in the study in group assignment or by sex. In addition, no significant attrition bias with respect to study measures was noted; those lost to follow-up did not differ from remaining participants on baseline RLS or depression scores. However, those who remained in the study were significantly older than those who dropped out.
Comparison of Those Who Completed the Study With Those Lost to Follow-up
Study Group Differences
Descriptive characteristics of study participants are presented in Table 3. The average age of participants was approximately 50 years (range, 18–72 years). Three quarters of the sample were women. Because of the study inclusion criteria, all participants had elevated IRLSS scores at baseline, with a total score range of 15 to 37. The 2 study groups did not differ significantly (P > .05) on demographic characteristics or baseline scores on any study measures.
Participant Descriptives by Study Group Assignment
Study group differences at 3 weeks are presented in Table 4. As described earlier, all analyses were intention to treat, and participants lost to follow-up were assigned their most recent previous score on each measure for analysis. Compared with those who received placebo, participants who took bupropion had significantly lower IRLSSG scores at 3 weeks, indicating symptom improvement. In addition, the average difference between baseline and 3-week IRLSS scores differed significantly between the 2 study groups, with those taking bupropion experiencing nearly an 11 point drop in total score. The 2 groups did not have significantly different scores on the depression measure at 3 weeks.
Three- and Six-Week Outcomes by Study Group Assignment*
Differences at 6 weeks also are presented in Table 4. At 6 weeks after baseline, the active drug group had maintained their decrease in total IRLSS scores and IRLSS differences from baseline that were evident at 3 weeks. However, the placebo group reported some additional improvement in RLS symptoms at 6 weeks. As a result, the study group differences in RLS symptoms found at 3 weeks, while trending in the expected direction, were no longer significant at 6 weeks.
Follow-up analyses were performed to determine if lack of medication compliance or elevated levels of depression may at least partially explain why the treatment effect seen at 3 weeks was no longer statistically significant at the 6-week assessment. No specific instructions had been given regarding compliance, and participants did not double their dosage in response to a missed dose. No verification of compliance other than participant self-reporting was obtained. Those who received bupropion were no more likely to skip pills than those who received the placebo (t(58) = 0.23; P = .760). In addition, on average, active drug group participants reported missing fewer than one pill per week (mean, 0.7). Finally, as shown in Table 4, placebo group participants reported a decrease in depression symptoms at 6 weeks compared with depression scores at 3 weeks. However, though the placebo group had somewhat lower depression scores at 6 weeks compared with the active drug group, because of small sample size these differences were not statistically significant, and further follow up analyses could not be performed.
Discussion
Bupropion improved the symptoms of RLS compared with placebo at 3 weeks. The difference at 6 weeks was not statistically significant because of improvements in the placebo group; however, the improvements in the bupropion group persisted through 6 weeks. The degree of improvement with bupropion was similar to the improvement seen with dopamine agonists currently approved for the treatment of moderate to severe RLS.10,11
In our study, bupropion did not exacerbate the symptoms of RLS. This contrasts with other antidepressants, which have been shown to exacerbate symptoms.7 As such, bupropion should be considered for depressed patients with RLS.
Our study did not definitively answer the question of whether bupropion might be an effective primary treatment for the symptoms of RLS. There was improvement at 3 weeks and a trend to continued improvement at 6 weeks. The improvement in symptoms was independent of the effect of bupropion on depression.
Strengths of our study included evaluation of a generic, relatively inexpensive medication with which family physicians have experience. Our entire minimal funding of $2,000 was provided by our university, and thus we received no industry support. In addition, randomization was appropriate, yielding similar groups at baseline. Finally, the degree of improvement in RLS symptoms among those who received bupropion in the current study is similar to improvements seen in studies of medications indicated for treatment of RLS.
Weaknesses of our study included the following. First, because of our limited resources, we were unable to recruit our target of 100 participants, which may have contributed to type II error at 6 weeks; that is, there may have been an effect that was not evident because of insufficient statistical power due to a small number of participants. Second, though analysis was by intention to treat, individuals were excluded from this analysis if they were inappropriately screened (see Results). However, all those with whom we were able to make contact and who actually began taking the medication were included in the analysis. Thus, the analysis did include all participants who chose to discontinue the medication for any reason. It also included all participants we were unable to reach or who failed to show up for a research appointment after the first week. We believe this is appropriate because, although intention to treat analysis may be defined to include all who were initially determined to be eligible, this is not essential. Rather, studies should be transparent and identify their definition of intention to treat as well as any effects the missing responses may have on the validity of the data.12 We feel the effect is minimal for the reasons mentioned above. Another weakness of our study was our relatively high drop-out rate. Reasons for withdrawing from the study are listed in Table 5. However, all who dropped out were included in the intention to treat analysis. An additional weakness is that we did not titrate the dose of bupropion to symptom improvement. The dose for all participants throughout the study was the starting dose of the sustained-release bupropion; it is possible that increasing the dose would have resulted in further improvement. It is also possible that it would have resulted in increased side effects. Our primary obstacle to titrating this dose was lack of funding.
Reasons for Withdrawing from Study
Conclusions
Our study demonstrates that bupropion does not exacerbate the symptoms of RLS. Furthermore, bupropion improved the symptoms of RLS at 3 weeks compared with placebo. The improvement persisted at 6 weeks, but further improvement among those who received the placebo resulted in lack of statistical significance, though there was a trend to improvement.
Further studies may clarify the role of bupropion in the treatment of RLS, and larger studies may determine if bupropion is effective as a primary treatment for the symptoms of moderate to severe RLS. Another important line of study would be to determine if adding bupropion to an SSRI would improve SSRI-induced symptoms of RLS. We did not exclude from our study those taking SSRIs, but because of small sample size, we were unable to analyze that group separately. Future studies of bupropion and RLS should include titration of dose to improvement. This is common in studies of dopamine agonists, but our limited resources and small sample size precluded this. Because our dose of sustained-release bupropion was the starting dose, it is possible that increasing the dose could result in greater improvement. With bupropion's reasonable cost and favorable side effect profile, it is our hope that some of these questions will be addressed in future studies.
Notes
This article was externally peer reviewed.
Funding: This study was funded in part by a grant from the East Tennessee State University Research Development Committee (MB) and in part by the Department of Family Medicine, East Tennessee State University.
Conflict of interest: none declared.
- Received for publication July 23, 2011.
- Revision received February 9, 2011.
- Accepted for publication February 14, 2011.



{kind=link}