Article Text

Abstract
Background: There have been many papers on the diagnostic criteria for specific hereditary cancer susceptibility syndromes and the likelihood that an individual has a germline mutation in one of the various cancer susceptibility genes. To assist health care professionals in deciding when a cancer genetics consultation is appropriate, available reports were critically reviewed in order to develop a single set of risk assessment criteria.
Methods: The criteria were based on a comprehensive review of publications describing diagnostic criteria for hereditary cancer syndromes and risk to first degree relatives of cancer patients. Priority was given to diagnostic criteria from consensus statements (for example, those from the National Comprehensive Cancer Network). Expert opinion from study personnel was then used to adopt a single set of criteria from other publications whenever guidelines differed.
Results: Based on family history, a set of criteria was developed to identify patients at risk for a hereditary cancer susceptibility syndrome, patients with moderate risk who might benefit from increased cancer surveillance, and patients who are at average risk. The criteria were applied to 4360 individuals who provided their cancer family history between July 1999 and April 2002, using a touch screen computer system in the lobby of a comprehensive cancer centre. They categorised an acceptable number of users into each risk level: 14.9% high risk, 13.7% moderate risk, and 59.6% average risk; 11.8% provided insufficient information for risk assessment.
Conclusions: These criteria should improve ease of referral and promote consistency across centres when evaluating patients for referral to cancer genetics specialists.
- family history
- risk assessment
- cancer
- heredity
- CRC, colorectal cancer
- FDR, first degree relative
- HBOC, hereditary breast and/or ovarian cancer syndrome
- HNPCC, hereditary non-polyposis colon cancer syndrome
- LFL, Li-Fraumeni-like
- LFS, Li-Fraumeni syndrome
- MEN, multiple endocrine neoplasia
- NCCN, National Comprehensive Cancer Network, SDR, second degree relative
Statistics from Altmetric.com
- CRC, colorectal cancer
- FDR, first degree relative
- HBOC, hereditary breast and/or ovarian cancer syndrome
- HNPCC, hereditary non-polyposis colon cancer syndrome
- LFL, Li-Fraumeni-like
- LFS, Li-Fraumeni syndrome
- MEN, multiple endocrine neoplasia
- NCCN, National Comprehensive Cancer Network, SDR, second degree relative
Health care providers have been encouraged to collect and analyse systematically the family histories of cancer in their patients, so as to facilitate prevention efforts and screening of relatives. Further, “duty to warn” litigation1 has underscored the importance of notifying cancer patients (and immediate family members) if they are at risk for a hereditary cancer susceptibility syndrome. This task usually involves, first, obtaining a cancer family history; second, determining whether a hereditary susceptibility exists; and third, communicating this risk assessment to patients and their families. To this end, health care providers are beginning to obtain cancer family histories from their patients in a concerted effort to provide risk assessment. Providers generally obtain information about first and second degree relatives affected by cancer, including the type of cancer and the age at diagnosis. For more information about taking a cancer family history, visit the National Cancer Institute Cancer Genetics PDQ at <http://www.cancer.gov/cancerinfo/pdq/genetics/risk-assessment-and-counseling#section_18>. Interpretation of these family history data, however, remains the greatest challenge to health care providers, who may not have expertise in the area of cancer genetics.2
A major limitation of cancer family risk assessment has been the variability of the risk assessment criteria used among institutions and individual clinicians. Many centres do not have written risk assessment criteria and rely on the expert opinion of the individual performing the assessment. In some cases, there are explicit criteria for the assessment of hereditary risk that may be missed by a limited review of the family history. For example, hereditary non-polyposis colon cancer syndrome (HNPCC) is an inherited cancer syndrome characterised primarily by colorectal and endometrial cancers. If the assessment during an annual gynaecological examination or mammogram is restricted to questions about breast or ovarian cancer, it is possible that HNPCC families may go undetected.
In order to facilitate and provide consistency in risk assessment, we sought to develop a set of criteria for use by clinicians gathering cancer family history information. The criteria are currently being used at two separate comprehensive cancer centres which collect family history data in different ways—one with a touch screen computer and the other with computer scanned forms. It would be ideal if these criteria could be used in a wide variety of settings by anyone collecting cancer family history information.
METHODS
Search strategy and selection criteria
A literature search was conducted using the MeSH headings “genetic predisposition to disease,” “genetic screening,” “neoplasms,” and “genetic counseling.” Risk assessment criteria differed from study to study and most only addressed a single hereditary cancer susceptibility syndrome. The papers identified underwent critical review with priority given to:
-
diagnostic criteria from consensus statements, for example the National Comprehensive Cancer Network, and so on;
-
research studies providing empirical data on the likelihood of having a mutation in a cancer susceptibility gene;
-
studies describing the characteristics of various hereditary cancer syndromes.
In cases where criteria differed among studies, the expert opinion of the authors was used to choose those least likely to miss patients with a hereditary cancer predisposition.
The risk assessment criteria were written to categorise individuals into three different risk levels.
High risk: Family histories suggestive of a hereditary cancer susceptibility syndrome where the individual would benefit from referral to a cancer genetics professional and increased cancer surveillance;
Moderate risk: Family histories not diagnostic of a hereditary cancer susceptibility syndrome but conferring an increased risk for cancer, requiring increased cancer surveillance;
Average risk: Family histories that do not require increased cancer surveillance and for whom the screening recommendations for the general population apply (that is, in the USA, those of the American Cancer Society).
The risk criteria are intended for use with both healthy at-risk individuals and cancer patients. If the individual providing their family history has had cancer, they should be counted as a first degree relative (FDR) affected with cancer when applying the risk assessment criteria. For example, if the user was diagnosed with breast cancer at age 39, they meet the criteria “FDR diagnosed with breast cancer under age 40 and should be categorised as high risk breast and referred for genetic counselling.”
Each risk category results in a different recommended intervention (fig 1). Individuals who meet the high risk criteria should be referred for clinical cancer genetics consultation and offered surveillance for the cancers associated with the suspected cancer predisposition syndrome. Cancer genetics professionals—including genetic counsellors, geneticists, oncologists, and nurses with experience in this area—can be found in the USA by visiting the National Cancer Institute website (http://www.cancer.gov/search/genetics_services/) or the National Society of Genetic Counselors website (http://www.nsgc.org), or by phoning the NCI information service at 1-800-4-CANCER.

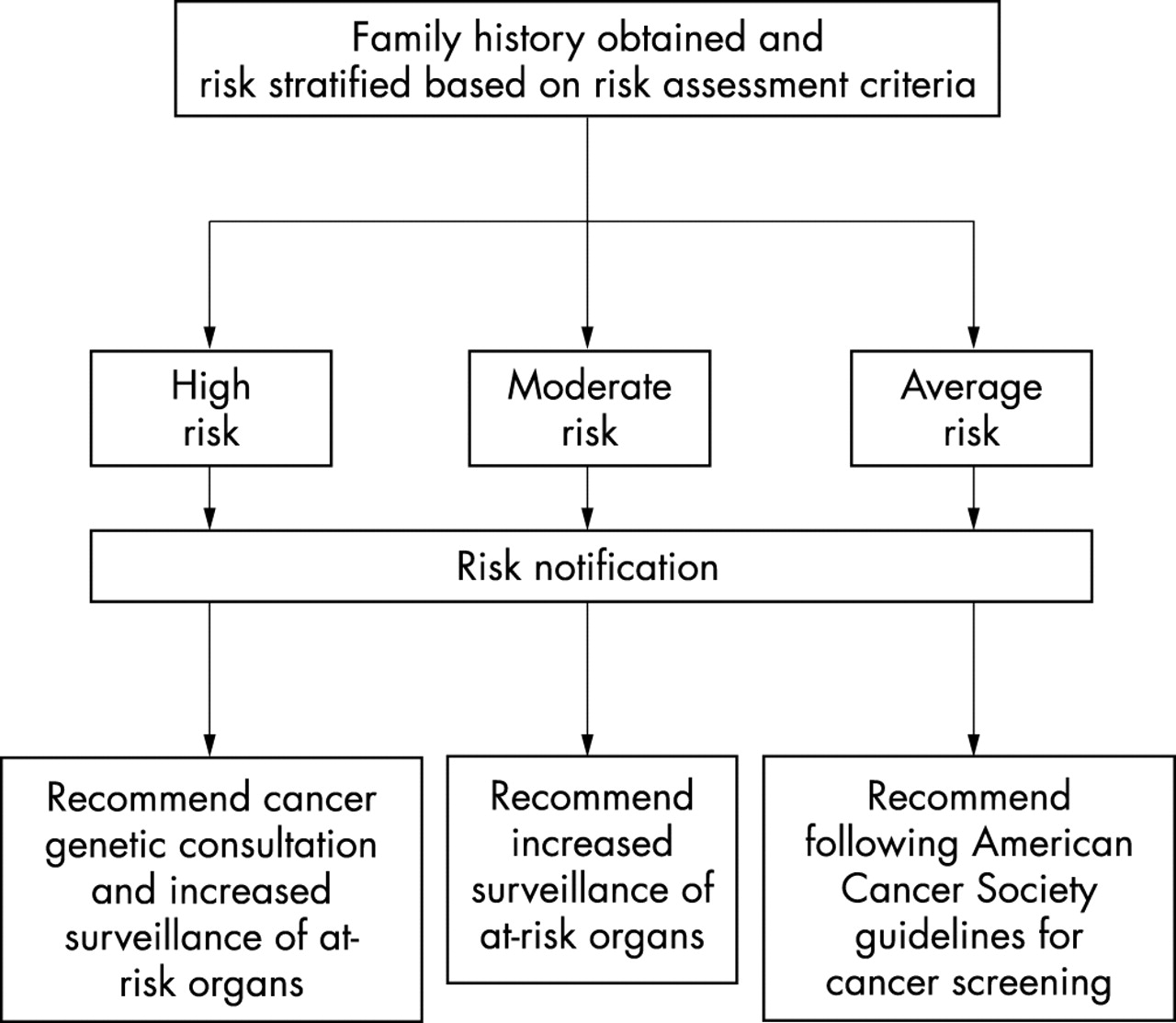{kind=link}
Flow chart for risk assessment and risk stratification.
The goal of the high risk criteria was to identify individuals or families that meet published diagnostic criteria for a particular cancer susceptibility syndrome or exceed a threshold of a 10% likelihood of finding a germline genetic mutation in a cancer susceptibility gene. This threshold is consistent with the American Society of Clinical Oncology statement3 that genetic testing might be offered to anyone who has a >10% chance of having a mutation in a cancer susceptibility gene. If the user or a first degree relative would have a >10% likelihood of having a mutation in a cancer susceptibility gene, we recommend referral for cancer genetics consultation. If the user is a healthy at-risk relative, they will learn in their consultation that genetic testing would best be initiated by a relative who has had cancer and probably has a greater chance of having a mutation.
While the criteria used to classify a family as high risk are based on the likelihood of having a cancer gene mutation, the outcome is referral for cancer genetic consultation and not testing, as some individuals will decide against testing for various reasons. Further, it is understood that some individuals with less than a 10% chance of having a cancer gene mutation would benefit from a cancer genetics consultation and these individuals could certainly seek out this service on their own. However, for the sake of large scale or high throughput risk assessment, the numbers of individuals being referred for genetic consultation must be limited in some way, and requiring a high likelihood of having a cancer gene mutation seems the most equitable.
The moderate risk criteria aim to identify individuals with a relative risk of ⩾2.0 for developing a particular cancer but who do not meet the specific criteria for a cancer predisposition syndrome. These individuals are at least twice as likely as someone in the general population to develop that particular cancer, and could benefit from increased cancer surveillance beyond that recommended for the general population. Consideration was given to the availability and efficacy of cancer surveillance procedures. Thus cancers omitted from the moderate risk criteria include pancreatic, lung, stomach, oesophagus, small intestine, brain, and haematological malignancies. It is recognised that there are screening methods for some of these cancers (for example, upper endoscopy for gastric cancer and spiral computed tomography for lung cancer detection); however, they are not of proven efficacy in moderate risk individuals. In addition, there are other cancers for which the screening guidelines for the general population (from resources such as the American Cancer Society) are also appropriate for individuals at twice the general population risk for developing the cancer. These include cervical, endometrial, and non-melanoma skin cancers.
Individuals who do not meet the high risk or moderate risk criteria, meet the average risk criteria by default. These individuals should follow guidelines for cancer screening in the general population (the US guidelines written by the American Cancer Society can be found on-line at http://www.cancer.org).
RESULTS
The review process resulted in a set of risk assessment criteria (table 1), with the rationale summarised below, by organ systems. In general, with rare exceptions, selection of high risk criteria reflect the known hallmarks of any hereditary cancer susceptibility syndrome—young age at diagnosis, multifocal tumours, bilaterality, presence of more than one associated cancer, and multiple affected members in a family.
Risk assessment criteria
Risk assessment criteria for breast and ovarian cancers
The genetic differential diagnosis for breast cancer includes hereditary breast and/or ovarian cancer syndrome (HBOC), Cowden syndrome, and Li-Fraumeni syndrome.4 In contrast, the genetic differential diagnosis for ovarian cancer includes HBOC and HNPCC. Germline mutations in BRCA1 or BRCA2 genes are associated with HBOC, believed to be the most common inherited breast cancer syndrome. There is an extensive literature on risk assessment criteria for breast and ovarian cancers. Professional society guidelines are based on empirical data regarding the likelihood that a particular family history can be attributed to a BRCA mutation; therefore these professional guidelines were used for the high risk criteria. Epidemiological studies estimating the risk that an individual will develop breast or ovarian cancer based on her family history were used to develop the moderate risk criteria.
High risk breast-ovarian cancer criteria (non-Jewish families)
Because of the high frequency of three specific BRCA1 and BRCA2 founder mutations among the Ashkenazim (with a 2.5% likelihood of having one of these mutations in the general Ashkenazi Jewish population), criteria for people of Jewish descent are reviewed separately (below).
There are several professional society guidelines that describe individuals at increased risk for HBOC. The National Comprehensive Cancer Network (NCCN)—an affiliation of 17 US cancer centres given comprehensive status by the US National Cancer Institute—has developed guidelines for genetic/familial high risk screening using a committee of cancer genetics experts from the member institutions.5 The guidelines are very similar to those developed by the Kaiser Permanente managed care organisation6 and the American College of Medical Genetics (ACMG) and New York State Department of Health7 (table 2).
Consensus guidelines for hereditary breast-ovarian cancer
Criteria for referral of solitary presentations of breast cancer rests on whether the a priori risk of finding a germline mutation is ⩾10% when diagnosed before a certain age. The main difference between the guidelines is cut off age at diagnosis: 30 years (Kaiser-Permanente), 40 years (NCCN), and 45 years (ACMG). The empirical data are variable, ranging from a 5.9% BRCA1 and BRCA2 mutation frequency in women diagnosed before the age of 368 to 9.4% of those diagnosed before 35.9 Mutation prevalence tables from the only laboratory providing clinical BRCA testing in the USA indicate that 7.6% of women with breast cancer under the age of 50 have a mutation, even if there is no family history of ovarian cancer or breast cancer under the age of 50.10 From these population based studies, an age cut off of 35 years may be appropriate given the proximity to a 10% a priori risk for mutation. However, in view of the fact that research studies often use detection technologies that may only be 60–90% sensitive,11 the mutation likelihood of those diagnosed under the age of 35 is likely to be falsely diminished and so an age cut off of 40 is adopted here.
In addition to young age at diagnosis, our high risk breast-ovarian criteria also consider the presence of multiple affected members in a family, ovarian cancer, male breast cancer, and bilateral breast cancer. The three Society guidelines recommend referral of individuals with three family members affected with breast or ovarian cancer or both (the NCCN requires at least one of the diagnoses to be ovarian cancer or a breast cancer that is diagnosed under the age of 50 or bilateral, while the others do not). The high risk breast-ovarian criteria presented here mirror the NCCN criteria.
Models based on data derived from multiple studies12–14 are available to estimate probabilities of detecting BRCA mutations depending on personal or family history of cancer. While these models are reasonable predictors of the likelihood that a patient has a BRCA mutation, they have to be used with professional judgment in the context of broad and in-depth knowledge of the cancer genetics literature. For example, the Couch model requires averaging the ages of breast cancer diagnoses in a given family and can only be used for families with at least two cases of breast cancer. Further, these models do not lend themselves to large scale screening efforts. As a result, we recommend that individuals familiar with their limitations apply these models.
Moderate risk breast cancer criteria (non-Jewish individuals)
The two most frequently used models for predicting lifetime risk for developing breast cancer are the Gail model from the breast cancer detection and demonstration project15–17 and the Claus data from the cancer and steroid hormone (CASH) study.18,19 As our risk assessment model is based solely on family history, we do not collect the information necessary to do the Gail risk calculation (age at menarche, parity, age at first childbirth, number of breast biopsies, and presence of atypia in these biopsies). We have elected to use the CASH data instead, as they incorporate more extensive information about family history and have been found to offer the most comprehensive assessment of family history for women with one or more first or second degree relatives with breast cancer.20,21 These data do not in any way reflect the likelihood that a particular individual has a BRCA1 or BRCA2 gene mutation. The Claus tables were used to create the moderate risk breast cancer criteria, targeting women with a relative risk of ⩾2.0 (or a lifetime risk of ⩾20%) for developing breast cancer. For women with a single first degree relative affected by breast cancer, the relative risk exceeded 2.0 only when the relative is diagnosed under the age of 30. As we classify anyone with a first or second degree relative with breast cancer diagnosed under the age of 40 as high risk, this criterion is not included among the moderate risk criteria. Women with two first degree relatives diagnosed with breast cancer had a lifetime risk exceeding 20% when one of these relatives was diagnosed under the age of 50, regardless of the age of diagnosis of the second one; however, this would be classified as high risk, so it was not included in the moderate risk category. If both first degree relatives were diagnosed between the ages of 51 and 60, the relative risk exceeded 2.0 and this would be considered a moderate risk. For women with a first degree relative and a maternal second degree relative (maternal aunt or maternal grandmother) affected by breast cancer, the relative risk for developing breast cancer exceeded 2.0 when the sum of the ages of both these relatives was ⩽118 (table 3). Review of the Claus tables for two second degree relatives and for the combination of one maternal first degree relative and one paternal second degree relative indicates that one of the relatives would need to be diagnosed under the age of 50 to exceed a relative risk of 2.0, thus meeting the high risk breast criteria. As a result, these criteria were not included in the moderate risk criteria.
Derivation of age formula for breast cancer moderate risk criteria (>20%) using Claus tables
Moderate risk ovarian cancer criteria (non-Jewish families)
In 1992, Kerlikowske et al reviewed all case–control studies of family history as a risk factor for ovarian cancer.22 They found that the relative risk for developing ovarian cancer among women with a first degree relative with ovarian cancer ranged from 1.9 to 3.6. As this meets our minimum relative risk of 2.0, this is the criterion adopted for moderate risk ovarian cancer. Women with a first degree relative with ovarian cancer who do not meet the high risk guidelines should discuss increased ovarian cancer surveillance with their physician. While the efficacy of ovarian cancer surveillance is unproven, some women at this level of risk might consider prophylactic oophorectomy.
High risk breast-ovarian cancer criteria (Jewish families)
There are three founder mutations in the BRCA genes found in 2.5% of the Ashkenazi Jewish population.23 As a result, a Jewish individual has a greater than 10% likelihood of having a BRCA mutation with less personal and family history of breast and ovarian cancer than a non-Jewish individual. Data from the New York breast cancer study indicate that 10.2% of Jewish women with breast cancer have one of the three founder mutations, regardless of family history.24 Thus the high risk breast-ovarian cancer risk assessment criteria differ for Jewish and non-Jewish individuals. The high risk criteria adopted for use with Jewish families are also taken from the NCCN guidelines (table 2). Given the extensive nature of the high risk breast-ovarian criteria for Jewish individuals, there is no moderate risk breast or ovarian cancer category.
Colon and other HNPCC associated cancer criteria
High risk HNPCC criteria
Because HNPCC is believed to be the most common hereditary cause of colon cancer, the general colon cancer risk criteria are built around established guidelines for its diagnosis (table 4). The International Consortium on Hereditary Nonpolyposis Colorectal Cancer (ICG-HNPCC) established a set of clinical diagnostic criteria, known as the Amsterdam criteria, to promote consistency among research studies.25 The Amsterdam criteria have been criticised for not taking into account the extracolonic cancers that are often associated with HNPCC. To resolve this problem, new clinical criteria (Amsterdam II criteria) were proposed26 to identify families that are very likely to have HNPCC. However, these criteria are not intended to serve as a guide to exclude families from cancer genetic consultation or mutation analysis. Therefore less restrictive criteria were adopted for the high risk colon category by extending the Amsterdam II criteria to include stomach, ovarian, and pancreatic cancers in the list of extracolonic cancers, because of data consistently associating them with HNPCC. Ureteric and renal pelvic cancers are also HNPCC associated cancers but are too specific to be included on most cancer family history questionnaires. “Kidney” cancer was accepted in lieu of these cancers for the purposes of mass screening. The exact subtype can be determined at subsequent interactions. Finally, for families with three cases of HNPCC associated cancers, we removed the age (one diagnosis under age 50) and multiple generation requirements in order to be more inclusive.
Hereditary non-polyposis colorectal cancer guidelines
Two additional criteria were adopted for the high risk HNPCC category. These include any family with a first degree relative diagnosed with colon cancer under the age of 50 or a first or second degree relative with two or more HNPCC associated cancers. This is based on the large population based HNPCC screening study in Finland.27 Using these criteria, 27 of the 28 patients with colorectal cancer found to carry HNPCC mutations in that study would have been identified. Moreover, the Bethesda guidelines suggest that further evaluation for HNPCC (through microsatellite instability testing) is warranted in any individual with more than one HNPCC associated cancer and for anyone diagnosed with colon cancer under the age of 45.28 The Bethesda criteria were recently found to have the highest sensitivity in detecting mutation positive HNPCC cases when compared with either of the two Amsterdam criteria.29 The American Gastroenterology Association adjusted the Bethesda criteria to suggest microsatellite instability for any colorectal cancer diagnosed under age 50, providing further support for this age cut off.30 At a recent follow up meeting (11–13 December 2002) for the revision of the Bethesda guidelines, it was decided to increase the age from 45 to 50. For the criteria presented here, the age of 50 was selected to maintain sensitivity while balancing the need to limit unnecessary “false positive” referrals. Around 8% of individuals diagnosed with colorectal cancer are under the age of 50 according to the population based series of colorectal cancer cases in Finland.31
Moderate risk colon cancer criteria
The population data regarding lifetime risks for colorectal cancer and not the likelihood of having an HNPCC gene mutation were reviewed to develop the moderate risk category. It has been found that an individual with one first degree relative diagnosed with colorectal cancer under the age of 45 has a relative risk of 3.7 to 6.4 for developing this condition.32,33 If the relative with colorectal cancer was diagnosed after the age of 45, the relative risk decreased to 1.8 to 2.7. For individuals with two first degree relatives diagnosed at any age, the relative risk was 5.7.33 Individuals with one first degree and one second degree relative diagnosed with colorectal cancer had a relative risk of 4.16.33 Our moderate risk criteria, therefore, include individuals with two first degree relatives or one first degree and one second degree relative with colorectal cancer, when the first degree relative is diagnosed at age 50 or greater (otherwise they would meet the high risk criteria).
High risk polyposis
It is advisable to include a question in all cancer family history collection tools to ascertain whether any family members have been diagnosed with polyposis (defined loosely as more than 10 colonic polyps) in their lifetime. Anyone answering in the affirmative should be referred for cancer genetics consultation. Pathology reports can be reviewed and a physical examination done to evaluate the family further for the possibility of familial adenomatous polyposis, MYH polyposis, Peutz-Jeghers syndrome, juvenile polyposis, mixed polyposis, or Cowden syndrome/Bannayan–Ruvalcaba–Riley syndrome.
Prostate cancer criteria
High risk prostate cancer criteria
Researchers are actively studying families with multiple cases of prostate cancer in a search for the genes responsible for hereditary prostate cancer. This search has been complicated by the discovery that many loci (HPCX,34PCAP,35 and CAPB36) are involved. To date, only two genes—HPC1/RNASEL37,38 and HPC2/ELAC239,40—have been isolated, but each accounts for very few hereditary cases. The high risk prostate criteria are based on the fact that families with more than three affected individuals are eligible for prostate cancer genetic research studies and could benefit from cancer genetics consultation. The relaxed criteria requiring two affected individuals (one diagnosed under the age of 60) are included in order to accommodate possible X linked inheritance patterns; these could limit the number of affected individuals because of the lack of male to male transmission and the unaffected status of obligate carrier mothers.
Moderate risk prostate cancer criteria
The moderate risk prostate criteria are based on population studies of prostate cancer risk. These studies suggest that first degree relatives of prostate cancer patients have a relative risk of 2.0 to 3.2 for developing the disease.41–43 Although prostate cancer is more common in African-Americans in the USA, the relative risk for developing prostate cancer when a first degree relative was affected was found to be similar among black and white individuals, so our risk assessment criteria do not take race into account.44 These relative risks infer that individuals with a single first degree relative affected by prostate cancer should meet the moderate risk criteria; however, we elected to add an age restriction so that individuals only meet this criterion if the first degree relative was diagnosed under the age of 60. In the USA, the American Cancer Society recommends that men begin prostate cancer screening at age 50. As it is generally agreed that surveillance should begin at least 10 years before the earliest diagnosis in the family, individuals would only need to begin prostate cancer screening early (before the age of 50) if they have a first degree relative diagnosed under the age of 60.
Steinberg et al also found a relative risk of 4.9 for individuals with two first degree relatives affected by prostate cancer, and 8.8 for individuals with one first degree and one second degree relative affected.42 Given these high relative risks (>2.0) for developing prostate cancer, the moderate risk criteria include both individuals with two first degree relatives with prostate cancer over the age of 60, and also those with one first degree and one second degree relative affected by prostate cancer over the age of 60.
Melanoma criteria
High risk melanoma criteria
A review by the Melanoma Genetics Consortium found that 20–40% of families with three or more affected first degree relatives have a mutation in the CDKN2A gene, while mutations were only found in 5% of families with two affected first degree relatives.45 Approximately 15% of individuals with multiple primary melanomas will be found to have a CDKN2A mutation.46 Based on these data, our high risk criteria require at least three relatives affected by melanoma in at least two generations, or any individual with multiple primaries. In addition, there is evidence that mutations in the CDKN2A gene also lead to an increased risk for pancreatic cancer.47,48 Accordingly, pancreatic cancers are included in the risk criteria.
Moderate risk melanoma criteria
Population studies have shown that individuals with one or more first degree relatives affected by melanoma have a relative risk of 2 to 3 for developing melanoma.49 Therefore increased surveillance is recommended for anyone with at least one first degree relative with melanoma. Increased surveillance for melanoma is non-invasive and includes more frequent clinical skin examinations, full body photography, and routine self examination, more frequent clinical skin examinations, and full body photography when indicated.
Li-fraumeni criteria
High risk Li-Fraumeni syndrome criteria
The classic diagnostic criteria for the Li-Fraumeni syndrome (LFS) are based on the original epidemiological studies of Li-Fraumeni kindreds.50–52 Families meeting these criteria have a 71% chance of having a germline TP53 mutation.53,54 It is quite rare for a family to meet the classic definition of LFS, however, so broader criteria were developed to ensure that all potential LFS families are detected. Li-Fraumeni-like (LFL) families have been defined in many different ways.53,55 Depending on the definition of LFL used, as many as 22% are found to have germline mutations in TP53. For these high risk Li-Fraumeni criteria, the NCCN LFL criteria were adopted.5 As it is unlikely that a family history questionnaire will identify the specific adrenal pathology associated with LFS (adrenocortical tumours), our criteria allow for any type of adrenal tumour. However, if there is a documented case of adrenocortical carcinoma, the family should be referred for cancer genetics consultation (below).
Multiple endocrine neoplasias
It is difficult to provide an assessment for the multiple endocrine neoplasia (MEN) syndromes on the basis of information obtained in a family history collection tool because of the importance of the histopathology of each of the component tumours. For example, it is reasonable to ask about a family history of thyroid cancer but one cannot assume that family members would know the specific histology (papillary, follicular, or medullary). Similarly, one cannot easily assess by questionnaire whether a pancreatic cancer is an adenocarcinoma or an islet cell tumour, or whether an adrenal tumour is a phaeochromocytoma or another subtype. In addition, many of the component tumours are not malignant—such as parathyroid hyperplasia and pituitary adenomas—so individuals may not include these diagnoses in a cancer history.
High risk MEN 1 criteria
As the diagnosis of MEN 1 is based on the presence of pancreatic islet cell tumours, pituitary adenomas, and parathyroid hyperplasia, the risk assessment criteria attempt to identify families with at least two of these features. MEN 1 has historically been diagnosed when two close relatives or a single individual have at least two of the principal clinical features.56,57 A cancer genetics consultation is appropriate for families with two cases of pancreatic (islet cell) cancer, parathyroid (hyperplasia), or pituitary adenoma in first degree or second degree relatives (both diagnoses can occur in the same person).
High risk thyroid cancer criteria (MEN 2 and familial non-medullary thyroid cancer)
The features of MEN 2 include medullary thyroid cancer, phaeochromocytomas, and parathyroid hyperplasia. If a family has a single case of medullary thyroid cancer they should be referred for cancer genetic consultation because the likelihood of having a RET gene mutation exceeds 10%.58 However, it would be unusual for individuals to be able to report the exact histology of a relative’s thyroid cancer. As the medullary histology is rare among all thyroid cancers, it would not be prudent to refer every person who had a single relative with “thyroid cancer” for cancer genetic consultation. For this reason, when one is unsure of the thyroid cancer histology, one additional feature is required for a family to be referred for a cancer genetics consultation. Even if the thyroid cancers in these families are non-medullary, they may be eligible for research studies aiming to identify the genes responsible for familial non-medullary thyroid cancer.
Moderate risk thyroid cancer criteria
It is known that individuals with a single first degree relative diagnosed with thyroid cancer have a relative risk of >2.0 for developing thyroid cancer regardless of subtype.59,60 These individuals may want to talk to their physician about increased thyroid cancer surveillance that might include annual physical examination with appropriate biochemical testing and thyroid ultrasound as indicated.
Single cases of cancer requiring cancer genetics consultation
There is evidence that a single individual diagnosed with medullary thyroid cancer.58 adrenocortical carcinoma,61,62 phaeochromocytoma,63 or paraganglioma (including carotid body tumours and glomus tumours)64 has a ⩾10% chance of having a hereditary cancer susceptibility syndrome. Therefore, in keeping with the other high risk categories, we recommend that individuals diagnosed with the above cancers, or those with a first degree relative diagnosed with these cancers, be referred for cancer genetics consultation regardless of their age at diagnosis or their family history.
Wilms’ tumour and retinoblastoma are known to be hereditary when bilateral or multifocal. As this is difficult to determine by questionnaire, one can either refer all individuals with these rare tumour types or risk missing some hereditary cases. Thus we have elected to suggest referral of all patients with a diagnosis of these tumours in themselves or a first degree relative for further consultation. Pathology reports can be reviewed at that time to determine whether or not the tumours are likely to be hereditary.
Familial aggregation of cancer
This broad category is critical for identifying families with cancer clusters suggestive of a hereditary predisposition. A cancer cluster can loosely be defined when a family has more cases of a particular cancer than one would expect to see by chance alone. To capture these clusters, the familial aggregation criteria state that any family with three cases (in first or second degree relatives) of the same malignancy on one side of the family should be considered high risk and be referred for a cancer genetics consultation. There are known cancer predisposition genes for some of these clusters—for example, basal cell naevus syndrome (PTCH), familial gastric cancer (CDH1), familial papillary renal cell carcinoma (MET), and renal cell cancers associated with Von Hippel Lindau disease (VHL) and Birt-Hogg-Dubé syndrome (BHD). For other cancer clusters, the responsible genes are yet to be identified. In these cases, families can be offered participation in research studies and familial tumour registries (that is, familial non-papillary renal cell carcinomas, familial pancreatic cancer, familial testicular cancer, familial oesophageal cancer, and familial haematological malignancies).
Other hereditary cancer predisposition syndromes
Cancer susceptibility syndromes not specifically addressed in these risk assessment criteria include Cowden syndrome, Von Hippel Lindau syndrome, tuberous sclerosis, neurofibromatosis, Carney complex, multiple osteochondromatosis, familial paraganglioma, Werner syndrome, and chromosome fragility syndromes (ataxia-telangiectasia, Bloom syndrome, Fanconi anaemia, and xeroderma pigmentosum). These syndromes include many non-cancer features and physical stigmata that are impossible to address in a large scale family history survey format. It is likely that families with Cowden syndrome could be identified by the high risk polyposis, thyroid, or breast-ovarian criteria.65 Some families with Von Hippel Lindau syndrome could be identified by a familial aggregation of renal cancers or apparently isolated presentations of phaeochromocytoma. In addition, the chromosome fragility and progerioid syndromes, Werner syndrome, and Rothmund–Thomson syndrome are usually autosomal recessive conditions where the diagnosis is made on the basis of dysmorphic features in the proband. Often only one family member is affected, making it difficult to detect these patients from their cancer family history.
Use of the risk assessment criteria
Two free standing family history risk assessment units using touch screen computer technology have been placed in the lobby of the James Cancer Hospital/Comprehensive Cancer Center and the JamesCare Dublin ambulatory care facility. Patients, family members, or passers by can use these machines on a voluntary basis to provide information about their personal cancer history and their family history of cancer in first and second degree relatives. Information on family history was provided by 4360 users between mid-1999 and April 2002. These risk assessment criteria were applied and users were categorised into high, moderate, and average risk groups. Of all users, 651 (14.9%) were found to be high risk, 598 (13.7%) were found to be moderate risk, and 2598 (59.6%) were found to be average risk, based on these criteria. As 5–10% of most cancers are thought to be hereditary (that is, high risk), while another 15% are familial (moderate risk), we feel that these criteria were validated in practice. It is important to note that 513 (11.8%) of the users provided insufficient information (generally unfinished entries) to assess risk. If these users all fell into the high risk category, it is possible that the criteria are too loose and lead to over-referral for cancer genetics consultation. However, in clinical practice, criteria which lead to “over-referral” are more conservative than those that miss true high risk cases. Nonetheless, the criteria will need to be validated in other settings to ensure wide scale applicability.
DISCUSSION
We have outlined the development of a set of criteria for use in assessing the cancer family history. An attempt was made to include all known hereditary cancer susceptibility syndromes in the development of risk assessment criteria. The criteria were developed for use with a computer assisted or paper family history questionnaire. There are limits to the information obtained by the usual collection tools that affect the ability to screen for certain syndromes (for example, some require a physical examination). Furthermore, most individuals cannot provide information on the exact histological subtypes of cancer. Risk assessment criteria must accommodate such limitations, always erring on the side of the highest risk scenario, so that families with hereditary cancer susceptibility syndromes are not missed. Practitioners who elicit a family history directly from an individual may be able to overcome the limitations.
These criteria do not address lifestyle and other epidemiological cancer risk factors. As a result, individuals are not given precise quantitative lifetime risks for cancer. Instead, they are placed in the general risk categories of high risk, moderate risk, and average risk, based only on their family history. The limitations of the risk assessment can be discussed in the risk notification communication process. In addition, family histories are dynamic and need to be updated regularly because additional relatives may be diagnosed with cancer since the last “risk assessment” occurred.
The importance of documentation of recommendations for both cancer genetics consultation and appropriate cancer surveillance based on family history assessment cannot be overemphasised. Identifying high risk families can save lives. For example, if a MEN 2A family is identified, counselling and genetic testing will lead to prophylactic thyroidectomy in mutation positive children in the family. This can prevent the development of medullary thyroid cancer.66 Likewise, there is evidence that beginning colonoscopies earlier and repeating them more often will dramatically reduce (and maybe eliminate) deaths from colon cancer in HNPCC families.67 This should apply to individuals meeting moderate risk colon cancer criteria as well.
There will not be 100% compliance with the recommendation for a cancer genetics consultation.68 Thus high risk patients must be alerted to the risk for all cancers associated with a hereditary cancer syndrome. This is especially important because many families with site specific cancers are not aware of the increased risks for other associated cancers.
While published reports have defined some of the hereditary cancer syndromes, risk assessment criteria vary and expert opinion was also critical in the development of these criteria. Thus formal molecular based validation is necessary. Large scale validation may best be undertaken by an organisation such as the NCCN or a large consortium. Ideally, molecular analysis of large populations should be used to determine the percentage of hereditary cancer syndrome families that would be identified by and missed by the high risk criteria.
Cancer risk assessment criteria will continue to evolve as the definitions of hereditary cancer susceptibility syndromes are further refined. Adoption and oversight by a national body would promote the timely revision and standardisation of the risk assessment criteria as new cancer syndromes are discovered and our current knowledge changes. As risk assessment is only as accurate as the family history provided, the importance of documentation is evident.
Increasing numbers of clinicians are striving to document adequate family history information to determine whether a hereditary cancer susceptibility exists, and to notify patients of their risk assessment. While the family history collection tools and means of risk notification may vary, the risk assessment criteria should be standardised. This review presents the first comprehensive evidence based risk assessment criteria for hereditary cancer syndromes, which constitute a first approximation of a uniform approach to familial cancer risk assessment that is usable in clinical practice.
Acknowledgments
We thank Albert de la Chapelle, Carrie Drovdlic, Rob Pilarski, and Rebecca Nagy for critical review of the manuscript. We are grateful to Karen Brown, Beth Peshkin, Charlene Schulz, and Lauren Scheuer, who worked on the early prototypes of cancer family history screening tools. This work was partially funded by National Cancer Institute grant P30CA16058 (to The Ohio State University Comprehensive Cancer Center) and generous donations from the Brown family in memory of Welton D Brown (to CE). CE is a recipient of the Doris Duke distinguished clinical scientist award.