Current Understanding of West Nile Virus Clinical Manifestations, Immune Responses, Neuroinvasion, and Immunotherapeutic Implications

{kind=link}
Abstract
:1. Introduction to West Nile virus
2. WNV Epidemiology
3. Clinical Manifestations of WNV Infection
4. Protective Immune Responses to WNV Infection
4.1. Innate Immunity
4.2. Complement
4.3. Adaptive Humoral Immunity
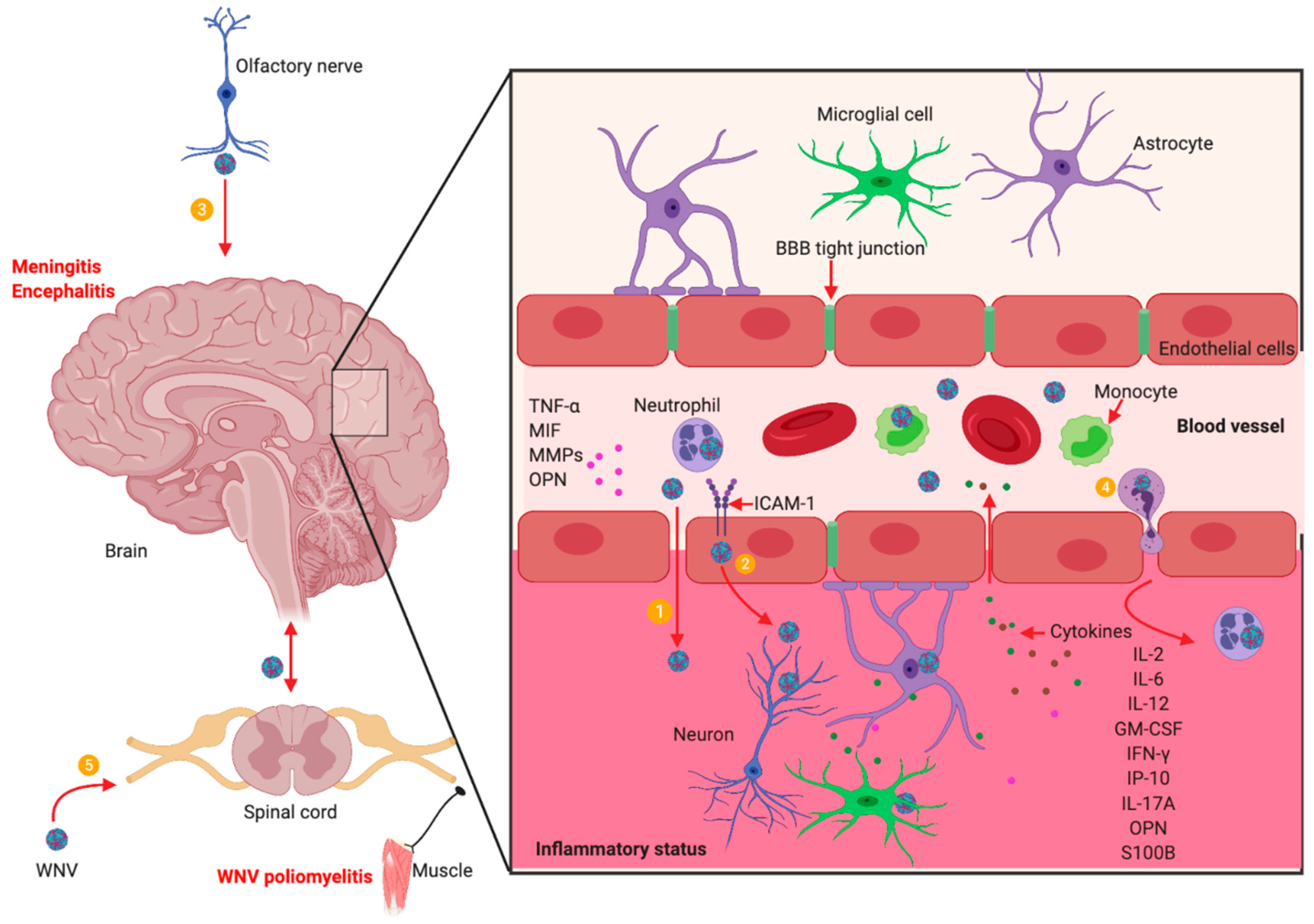
5. WNV Neuroinvasion and Neuropathogenesis
6. Immunotherapeutic Intervention
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Martin-Acebes, M.A.; Saiz, J.C. West Nile virus: A re-emerging pathogen revisited. World J. Virol. 2012, 1, 51–70. [Google Scholar] [CrossRef]
- Hackett, B.A.; Cherry, S. Flavivirus internalization is regulated by a size-dependent endocytic pathway. Proc. Natl. Acad. Sci. USA 2018, 115, 4246–4251. [Google Scholar] [CrossRef] [Green Version]
- Ng, W.C.; Soto-Acosta, R.; Bradrick, S.S.; Garcia-Blanco, M.A.; Ooi, E.E. The 5′ and 3′ Untranslated Regions of the Flaviviral Genome. Viruses 2017, 9, 137. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, W.; Li, J.; Wu, W.; Jiu, Y. The Role of Host Cytoskeleton in Flavivirus Infection. Virol. Sin. 2019, 34, 30–41. [Google Scholar] [CrossRef]
- Mazeaud, C.; Freppel, W.; Chatel-Chaix, L. The Multiples Fates of the Flavivirus RNA Genome During Pathogenesis. Front. Genet. 2018, 9, 595. [Google Scholar] [CrossRef]
- EFirth, A.; Atkins, J.F. A conserved predicted pseudoknot in the NS2A-encoding sequence of West Nile and Japanese encephalitis flaviviruses suggests NS1’ may derive from ribosomal frameshifting. Virol. J. 2009, 6, 14. [Google Scholar] [CrossRef]
- Young, L.B.; Melian, E.B.; Setoh, Y.X.; Young, P.R.; Khromykh, A.A. Last 20 aa of the West Nile virus NS1′ protein are responsible for its retention in cells and the formation of unique heat-stable dimers. J. Gen. Virol. 2015, 96, 1042–1054. [Google Scholar] [CrossRef]
- Colpitts, T.M.; Conway, M.J.; Montgomery, R.R.; Fikrig, E. West Nile Virus: Biology, Transmission, and Human Infection. Clin. Microbiol. Rev. 2012, 25, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Spigland, I.; Jasinska-Klingberg, W.; Hofshi, E.; Goldblum, N. Clinical and laboratory observations in an outbreak of West Nile fever in Israel in 1957. Harefuah 1958, 54, 275–280. [Google Scholar]
- Weinberger, M.; Pitlik, S.D.; Gandacu, D.; Lang, R.; Nassar, F.; Ben David, D.; Rubinstein, E.; Izthaki, A.; Mishal, J.; Kitzes, R.; et al. West Nile fever outbreak, Israel, 2000: Epidemiologic aspects. Emerg. Infect. Dis. 2001, 7, 686–691. [Google Scholar] [CrossRef]
- Campbell, G.L.; Marfin, A.A.; Lanciotti, R.S.; Gubler, D.J. West Nile virus. Lancet Infect. Dis. 2002, 2, 519–529. [Google Scholar] [CrossRef]
- Mostashari, F.; Fine, A.; O’Leary, D.; Huang, A.; Sherman, M.; Wong, S.; Campbell, G.L.; Shieh, W.-J.; Smith, P.; Greenberg, A.; et al. The Outbreak of West Nile Virus Infection in the New York City Area in 1999. N. Engl. J. Med. 2001, 344, 1807–1814. [Google Scholar]
- Chancey, C.; Grinev, A.; Volkova, E.; Rios, M. The Global Ecology and Epidemiology of West Nile Virus. Biomed Res. Int. 2015, 2015, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Kramer, L.D.; Li, J.; Shi, P.Y. West Nile virus. Lancet. Neurol. 2007, 6, 171–181. [Google Scholar] [CrossRef]
- Kaiser, J.A.; Barrett, A.D. Twenty Years of Progress toward West Nile Virus Vaccine Development. Viruses 2019, 11, 823. [Google Scholar] [CrossRef]
- CDC. West Nile Virus. Available online: https://www.cdc.gov/westnile/healthcareproviders/healthcareproviders-diagnostic.html (accessed on 10 December 2018).
- Martinovic, V.; Kisic-Tepavcevic, D.; Kacar, A.; Mesaros, S.; Pekmezovic, T.; Drulovic, J. Longitudinally extensive transverse myelitis in a patient infected with West Nile virus. Mult. Scler. Relat. Disord. 2019, 32, 19–22. [Google Scholar] [CrossRef]
- Pradhan, S.; Anand, S.; Choudhury, S.S. Cognitive behavioural impairment with irreversible sensorineural deafness as a complication of West Nile encephalitis. J. Neurovirol. 2019, 25, 429–433. [Google Scholar] [CrossRef]
- Burdmann, E.A. Flaviviruses and Kidney Diseases. Adv. Chronic Kidney Dis. 2019, 26, 198–206. [Google Scholar] [CrossRef]
- Conetta, R.; Teixeira, A.A.; Waldman, G.; Sampson, B.A.; Asnis, D.S. The West Nile Virus Outbreak of 1999 in New York: The Flushing Hospital Experience. Clin. Infect. Dis. 2000, 30, 413–418. [Google Scholar] [Green Version]
- Sampson, B.A.; Ambrosi, C.; Charlot, A.; Reiber, K.; Veress, J.F.; Armbrustmacher, V. The pathology of human West Nile virus infection. Hum. Pathol. 2000, 31, 527–531. [Google Scholar] [CrossRef]
- Leis, A.A.; Polk, J.L.; Dostrow, V.; Winkelmann, M.; Stokic, D.S. A Poliomyelitis-like Syndrome from West Nile Virus Infection. N. Engl. J. Med. 2002, 347, 1279–1280. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Loeb, J.A.; Shy, M.E.; Shah, A.K.; Tselis, A.C.; Kupski, W.J.; Lewis, R.A. Asymmetric flaccid paralysis: A neuromuscular presentation of West Nile virus infection. Ann. Neurol. 2003, 53, 703–710. [Google Scholar] [CrossRef]
- Jeha, L.E.; Sila, C.A.; Lederman, R.J.; Prayson, R.A.; Isada, C.M.; Gordon, S.M. West Nile virus infection: A new acute paralytic illness. Neurology 2003, 61, 55–59. [Google Scholar] [CrossRef]
- Leis, A.A.; Stokic, D.S. Neuromuscular Manifestations of West Nile Virus Infection. Front. Neurol. 2012, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Leis, A.A.; Stokic, D.S.; Webb, R.M.; Slavinski, S.A.; Fratkin, J. Clinical spectrum of muscle weakness in human West Nile virus infection. Muscle Nerve 2003, 28, 302–308. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Leis, A.A.; Stokic, D.S.; Van Gerpen, J.A.; Marfin, A.A.; Webb, R.; Haddad, M.B.; Tierney, B.C.; Slavinski, S.A.; Polk, J.L.; et al. Acute Flaccid Paralysis and West Nile Virus Infection. Emerg. Infect. Dis. 2003, 9, 788–793. [Google Scholar] [CrossRef]
- Johnstone, J.; Hanna, S.E.; Nicolle, L.E.; Drebot, M.A.; Neupane, B.; Mahony, J.B.; Loeb, M.B. Prognosis of West Nile virus associated acute flaccid paralysis: A case series. J. Med. Case Rep. 2011, 5, 395. [Google Scholar] [CrossRef]
- Fratkin, J.D.; Leis, A.A.; Stokic, D.S.; Slavinski, S.A.; Geiss, R.W. Spinal cord neuropathology in human West Nile virus infection. Arch. Pathol. Lab. Med. 2004, 128, 533–537. [Google Scholar]
- Garcia, M.N.; Hause, A.M.; Walker, C.M.; Orange, J.S.; Hasbun, R.; Murray, K.O. Evaluation of Prolonged Fatigue Post–West Nile Virus Infection and Association of Fatigue with Elevated Antiviral and Proinflammatory Cytokines. Viral Immunol. 2014, 27, 327–333. [Google Scholar] [CrossRef]
- Pepperell, C.; Rau, N.; Krajden, S.; Kern, R.; Humar, A.; Mederski, B.; Simor, A.; Low, D.E.; McGeer, A.; Mazzulli, T.; et al. West Nile virus infection in 2002: Morbidity and mortality among patients admitted to hospital in southcentral Ontario. Can. Med. Assoc. J. 2003, 168, 1399–1405. [Google Scholar]
- Lindsey, N.P.; Sejvar, J.J.; Bode, A.V.; Pape, W.J.; Campbell, G.L. Delayed Mortality in a Cohort of Persons Hospitalized with West Nile Virus Disease in Colorado in 2003. Vector Borne Zoonotic Dis. 2012, 12, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Siddharthan, V.; Hall, J.O.; Morrey, J.D. Autonomic Nervous Dysfunction in Hamsters Infected with West Nile Virus. PLoS ONE 2011, 6, e19575. [Google Scholar] [CrossRef] [PubMed]
- Morrey, J.D.; Siddharthan, V.; Wang, H.; Hall, J.O.; Motter, N.E.; Skinner, R.D.; Skirpstunas, R.T. Neurological suppression of diaphragm electromyographs in hamsters infected with West Nile virus. J. Neurovirol. 2010, 16, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zukor, K.; Wang, H.; Hurst, B.L.; Siddharthan, V.; Van Wettere, A.; Pilowsky, P.M.; Morrey, J.D. Phrenic nerve deficits and neurological immunopathology associated with acute West Nile virus infection in mice and hamsters. J. Neurovirol. 2017, 23, 186–204. [Google Scholar] [CrossRef] [PubMed]
- Budhram, A.; Sharma, M.; Shettar, B.; Hosseini-Moghaddam, S.M.; Khaw, A.V. Sensory and autonomic involvement in West Nile virus-associated acute flaccid paralysis. Neurol. Clin. Pract. 2017, 7, 394–397. [Google Scholar] [CrossRef]
- Town, T.; Jeng, D.; Alexopoulou, L.; Tan, J.; Flavell, R.A. Microglia recognize double-stranded RNA via TLR3. J. Immunol. 2006, 176, 3804–3812. [Google Scholar] [CrossRef]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Gale, M.; Diamond, M.S. Toll-Like Receptor 3 Has a Protective Role against West Nile Virus Infection. J. Virol. 2008, 82, 10349–10358. [Google Scholar] [CrossRef] [Green Version]
- Scholle, F.; Mason, P.W. West Nile virus replication interferes with both poly(I:C)-induced interferon gene transcription and response to interferon treatment. Virology 2005, 342, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef]
- Town, T.; Bai, F.; Wang, T.; Kaplan, A.T.; Qian, F.; Montgomery, R.R.; Anderson, J.F.; Flavell, R.A.; Fikrig, E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 2009, 30, 242–253. [Google Scholar] [CrossRef]
- Liu, J.; Xu, C.; Hsu, L.C.; Luo, Y.; Xiang, R.; Chuang, T.H. A five-amino-acid motif in the undefined region of the TLR8 ectodomain is required for species-specific ligand recognition. Mol. Immunol. 2010, 47, 1083–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, A.M.; Acharya, D.; Le, L.; Wang, P.; Stokic, D.S.; Leis, A.A.; Alexopoulou, L.; Town, T.; Flavell, R.A.; Fikrig, E.; et al. TLR8 Couples SOCS-1 and Restrains TLR7-Mediated Antiviral Immunity, Exacerbating West Nile Virus Infection in Mice. J. Immunol. 2016, 197, 4425–4435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid–inducible gene-I and melanoma differentiation–associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Takahasi, K.; Kumeta, H.; Tsuduki, N.; Narita, R.; Shigemoto, T.; Hirai, R.; Yoneyama, M.; Horiuchi, M.; Ogura, K.; Fujita, T.; et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: Identification of the RNA recognition loop in RIG-I-like receptors. J. Biol. Chem. 2009, 284, 17465–17474. [Google Scholar] [CrossRef]
- Bruns, A.M.; Leser, G.P.; Lamb, R.A.; Horvath, C.M. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 2014, 55, 771–781. [Google Scholar] [CrossRef]
- Komuro, A.; Horvath, C.M. RNA- and Virus-Independent Inhibition of Antiviral Signaling by RNA Helicase LGP2. J. Virol. 2006, 80, 12332–12342. [Google Scholar] [CrossRef] [Green Version]
- Rothenfusser, S.; Goutagny, N.; DiPerna, G.; Gong, M.; Monks, B.G.; Schoenemeyer, A.; Yamamoto, M.; Akira, S.; Fitzgerald, K.A. The RNA Helicase Lgp2 Inhibits TLR-Independent Sensing of Viral Replication by Retinoic Acid-Inducible Gene-I. J. Immunol. 2005, 175, 5260–5268. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Hirai, R.; Loo, Y.M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Kato, H.; Kumagai, Y.; Yoneyama, M.; Sato, S.; Matsushita, K.; Tsujimura, T.; Fujita, T.; Akira, S.; Takeuchi, O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 2010, 107, 1512–1517. [Google Scholar] [CrossRef]
- Venkataraman, T.; Valdes, M.; Elsby, R.; Kakuta, S.; Caceres, G.; Saijo, S.; Iwakura, Y.; Barber, G.N. Loss of DExD/H Box RNA Helicase LGP2 Manifests Disparate Antiviral Responses. J. Immunol. 2007, 178, 6444–6455. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Ramos, H.J.; Brassil, M.M.; Netland, J.; Chappell, C.P.; Blahnik, G.; McMillan, A.; Diamond, M.S.; Clark, E.A.; Bevan, M.J.; et al. The RIG-I-like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity 2012, 37, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Diamond, M.S. Alpha/Beta Interferon Protects against Lethal West Nile Virus Infection by Restricting Cellular Tropism and Enhancing Neuronal Survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Wang, X.J.; Clark, D.C.; Lobigs, M.; Hall, R.A.; Khromykh, A.A. A Single Amino Acid Substitution in the West Nile Virus Nonstructural Protein NS2A Disables Its Ability To Inhibit Alpha/Beta Interferon Induction and Attenuates Virus Virulence in Mice. J. Virol. 2006, 80, 2396–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.R.; de Sessions, P.F.; Leon, M.A.; Scholle, F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J. Virol. 2008, 82, 8262–8271. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Gale, M. West Nile Virus Evades Activation of Interferon Regulatory Factor 3 through RIG-I-Dependent and -Independent Pathways without Antagonizing Host Defense Signaling. J. Virol. 2006, 80, 2913–2923. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.-T.; Hayashi, J.; Seeger, C. West Nile Virus Inhibits the Signal Transduction Pathway of Alpha Interferon. J. Virol. 2005, 79, 1343–1350. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Jordán, J.L.; Laurent-Rolle, M.; Ashour, J.; Martínez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; Garcia-Sastre, A. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef]
- Liu, W.J.; Chen, H.B.; Wang, X.J.; Huang, H.; Khromykh, A.A. Analysis of Adaptive Mutations in Kunjin Virus Replicon RNA Reveals a Novel Role for the Flavivirus Nonstructural Protein NS2A in Inhibition of Beta Interferon Promoter-Driven Transcription. J. Virol. 2004, 78, 12225–12235. [Google Scholar] [CrossRef] [Green Version]
- Setoh, Y.X.; Periasamy, P.; Peng, N.Y.G.; Amarilla, A.A.; Slonchak, A.; Khromykh, A.A. Helicase Domain of West Nile Virus NS3 Protein Plays a Role in Inhibition of Type I Interferon Signalling. Viruses 2017, 9, 326. [Google Scholar] [CrossRef]
- Ambrose, R.L.; Mackenzie, J.M. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 Protein of the Virulent West Nile Virus NY99 Strain Is a Potent Antagonist of Type I Interferon-Mediated JAK-STAT Signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan-Tack, K.M.; Forrest, G. Failure of interferon alpha-2b in a patient with West Nile virus meningoencephalitis and acute flaccid paralysis. Scand. J. Infect. Dis. 2005, 37, 944–946. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.M.; King, N.J. Accelerated dendritic cell differentiation from migrating Ly6C(lo) bone marrow monocytes in early dermal West Nile virus infection. J. Immunol. 2011, 186, 2382–2396. [Google Scholar] [CrossRef]
- Kovats, S.; Turner, S.; Simmons, A.; Powe, T.; Chakravarty, E.; Alberola-Ila, J. West Nile virus-infected human dendritic cells fail to fully activate invariant natural killer T cells. Clin. Exp. Immunol. 2016, 186, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Wang, X.; Zhang, L.; Lin, A.; Zhao, H.; Fikrig, E.; Montgomery, R.R. Impaired Interferon Signaling in Dendritic Cells From Older Donors Infected In Vitro With West Nile Virus. J. Infect. Dis. 2011, 203, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Martina, B.E.; Koraka, P.; van den Doel, P.; Rimmelzwaan, G.F.; Haagmans, B.L.; Osterhaus, A.D. DC-SIGN enhances infection of cells with glycosylated West Nile virus in vitro and virus replication in human dendritic cells induces production of IFN-alpha and TNF-alpha. Virus Res. 2008, 135, 64–71. [Google Scholar] [CrossRef]
- Silva, M.C.; Guerrero-Plata, A.; Gilfoy, F.D.; Garofalo, R.P.; Mason, P.W. Differential Activation of Human Monocyte-Derived and Plasmacytoid Dendritic Cells by West Nile Virus Generated in Different Host Cells. J. Virol. 2007, 81, 13640–13648. [Google Scholar] [CrossRef] [Green Version]
- Schmid, E.T.; Pang, I.K.; Silva, E.A.C.; Bosurgi, L.; Miner, J.J.; Diamond, M.S.; Iwasaki, A.; Rothlin, C.V. AXL receptor tyrosine kinase is required for T cell priming and antiviral immunity. eLife 2016, 5, e12414. [Google Scholar] [CrossRef]
- Aarreberg, L.D.; Wilkins, C.; Ramos, H.J.; Green, R.; Davis, M.A.; Chow, K.; Gale, M., Jr. Interleukin-1beta Signaling in Dendritic Cells Induces Antiviral Interferon Responses. mBio 2018, 9, e00342-18. [Google Scholar] [CrossRef]
- Chow, K.T.; Driscoll, C.; Loo, Y.M.; Knoll, M.; Gale, M., Jr. IRF5 regulates unique subset of genes in dendritic cells during West Nile virus infection. J. Leukoc. Biol. 2019, 105, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Lancaster, A.; Wilkins, C.; Suthar, M.S.; Huang, A.; Vick, S.C.; Clepper, L.; Thackray, L.; Brassil, M.M.; Virgin, H.W.; et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Ben-Nathan, D.; Huitinga, I.; Lustig, S.; Van Rooijen, N.; Kobiler, D. West Nile virus neuroinvasion and encephalitis induced by macrophage depletion in mice. Arch. Virol. 1996, 141, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Winkelmann, E.R.; Zhu, S.; Ru, W.; Mays, E.; Silvas, J.A.; Vollmer, L.L.; Gao, J.; Peng, B.-H.; Bopp, N.E.; et al. Peli1 facilitates virus replication and promotes neuroinflammation during West Nile virus infection. J. Clin. Investig. 2018, 128, 4980–4991. [Google Scholar] [CrossRef]
- Drescher, B.; Bai, F. Neutrophil in viral infections, friend or foe? Virus Res. 2013, 171, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, F.; Kong, K.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgometry, R.R. A Paradoxical Role for Neutrophils in the Pathogenesis of West Nile Virus. J. Infect. Dis. 2010, 202, 1804–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, A.M.; Acharya, D.; Duty, L.; Thompson, E.A.; Le, L.; Stokic, D.S.; Leis, A.A.; Bai, F. Osteopontin facilitates West Nile virus neuroinvasion via neutrophil “Trojan horse” transport. Sci. Rep. 2017, 7, 4722. [Google Scholar] [CrossRef]
- Wang, P.; Bai, F.; Zenewicz, L.A.; Dai, J.; Gate, D.; Cheng, G.; Yang, L.; Qian, F.; Yuan, X.; Montgomery, R.R.; et al. IL-22 Signaling Contributes to West Nile Encephalitis Pathogenesis. PLoS ONE 2012, 7, e44153. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Liu, Y.; Müllbacher, A. Astrocytes are not susceptible to lysis by natural killer cells. J. Neuroimmunol. 1988, 19, 101–110. [Google Scholar] [CrossRef]
- Shrestha, B.; Samuel, M.A.; Diamond, M.S. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J. Virol. 2006, 80, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Daniel, S.; Huang, Y.; Chancey, C.; Huang, Q.; Lei, Y.F.; Grinev, A.; Mostowski, H.; Rios, M.; Dayton, A. Anti-West Nile virus activity of in vitro expanded human primary natural killer cells. BMC Immunol. 2010, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Strauss-Albee, D.M.; Zhou, J.Q.; Malawista, A.; Garcia, M.N.; Murray, K.O.; Blish, C.A.; Montgomery, R.R. The natural killer cell response to West Nile virus in young and old individuals with or without a prior history of infection. PLoS ONE 2017, 12, e0172625. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Wang, T.; Samuel, M.A.; Whitby, K.; Craft, J.; Fikrig, E.; Diamond, M.S. Gamma Interferon Plays a Crucial Early Antiviral Role in Protection against West Nile Virus Infection. J. Virol. 2006, 80, 5338–5348. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Scully, E.; Yin, Z.; Kim, J.H.; Wang, S.; Yan, J.; Mamula, M.; Anderson, J.F.; Craft, J.; Fikrig, E. IFN-gamma-producing gamma delta T cells help control murine West Nile virus infection. J. Immunol. 2003, 171, 2524–2531. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Lamb, J.; Anderson, J.F.; Born, W.K.; O’Brien, R.L.; Wang, T. Role of two distinct gammadelta T cell subsets during West Nile virus infection. FEMS Immunol. Med. Microbiol. 2008, 53, 275–283. [Google Scholar] [CrossRef]
- Bai, F.; Town, T.; Qian, F.; Wang, P.; Kamanaka, M.; Connolly, T.M.; Gate, D.; Montgomery, R.R.; Flavell, R.A.; Fikrig, E. IL-10 Signaling Blockade Controls Murine West Nile Virus Infection. PLoS Pathog. 2009, 5, e1000610. [Google Scholar] [CrossRef]
- Welte, T.; Aronson, J.; Gong, B.; Rachamallu, A.; Mendell, N.; Tesh, R.; Paessler, S.; Born, W.K.; O’Brien, R.L.; Wang, T. Vgamma4+ T cells regulate host immune response to West Nile virus infection. FEMS Immunol. Med. Microbiol. 2011, 63, 183–192. [Google Scholar] [CrossRef]
- Mehlhop, E.; Whitby, K.; Oliphant, T.; Marri, A.; Engle, M.; Diamond, M.S. Complement Activation Is Required for Induction of a Protective Antibody Response against West Nile Virus Infection. J. Virol. 2005, 79, 7466–7477. [Google Scholar] [CrossRef] [Green Version]
- Mehlhop, E.; Diamond, M.S. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J. Exp. Med. 2006, 203, 1371–1381. [Google Scholar] [CrossRef]
- Fuchs, A.; Lin, T.-Y.; Beasley, D.W.; Stover, C.M.; Schwaeble, W.J.; Pierson, T.C.; Diamond, M.S. Direct complement restriction of flavivirus infection requires glycan recognition by mannose-binding lectin. Cell Host Microbe 2010, 8, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Liszewski, M.K.; Nybakken, G.; Davis, A.E.; Townsend, R.R.; Fremont, D.H.; Atkinson, J.P.; Diamond, M.S. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc. Natl. Acad. Sci. USA 2006, 103, 19111–19116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avirutnan, P.; Fuchs, A.; Hauhart, R.E.; Somnuke, P.; Youn, S.; Diamond, M.S.; Atkinson, J.P. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J. Exp. Med. 2010, 207, 793–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avirutnan, P.; Hauhart, R.E.; Somnuke, P.; Blom, A.M.; Diamond, M.S.; Atkinson, J.P. Binding of flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J. Immunol. 2011, 187, 424–433. [Google Scholar] [CrossRef]
- Mehlhop, E.; Nelson, S.; Jost, C.A.; Gorlatov, S.; Johnson, S.; Fremont, D.H.; Diamond, M.S.; Pierson, T.C. Complement protein C1q reduces the stoichiometric threshold for antibody-mediated neutralization of West Nile virus. Cell Host Microbe 2009, 6, 381–391. [Google Scholar] [CrossRef]
- Fuchs, A.; Pinto, A.K.; Schwaeble, W.J.; Diamond, M.S. The lectin pathway of complement activation contributes to protection from West Nile virus infection. Virology 2011, 412, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehlhop, E.; Fuchs, A.; Engle, M.; Diamond, M.S. Complement modulates pathogenesis and antibody-dependent neutralization of West Nile virus infection through a C5-independent mechanism. Virology 2009, 393, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B Cells and Antibody Play Critical Roles in the Immediate Defense of Disseminated Infection by West Nile Encephalitis Virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar] [CrossRef] [Green Version]
- Engle, M.J.; Diamond, M.S. Antibody Prophylaxis and Therapy against West Nile Virus Infection in Wild-Type and Immunodeficient Mice. J. Virol. 2003, 77, 12941–12949. [Google Scholar] [CrossRef]
- Diamond, M.S.; Sitati, E.M.; Friend, L.D.; Higgs, S.; Shrestha, B.; Engle, M. A Critical Role for Induced IgM in the Protection against West Nile Virus Infection. J. Exp. Med. 2003, 198, 1853–1862. [Google Scholar] [CrossRef]
- Xia, J.; Winkelmann, E.R.; Gorder, S.R.; Mason, P.W.; Milligan, G.N. TLR3- and MyD88-Dependent Signaling Differentially Influences the Development of West Nile Virus-Specific B Cell Responses in Mice following Immunization with RepliVAX WN, a Single-Cycle Flavivirus Vaccine Candidate. J. Virol. 2013, 87, 12090–12101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purtha, W.E.; Chachu, K.A.; Virgin, H.W.; Diamond, M.S. Early B-Cell Activation after West Nile Virus Infection Requires Alpha/Beta Interferon but Not Antigen Receptor Signaling. J. Virol. 2008, 82, 10964–10974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, M.J.; Poddar, S.; Farzan, M.; Diamond, M.S. The Interferon-Stimulated Gene Ifitm3 Restricts West Nile Virus Infection and Pathogenesis. J. Virol. 2016, 90, 8212–8225. [Google Scholar] [CrossRef] [PubMed]
- Thackray, L.B.; Shrestha, B.; Richner, J.M.; Miner, J.J.; Pinto, A.K.; LaZear, H.M.; Gale, M.; Diamond, M.S. Interferon Regulatory Factor 5-Dependent Immune Responses in the Draining Lymph Node Protect against West Nile Virus Infection. J. Virol. 2014, 88, 11007–11021. [Google Scholar] [CrossRef] [Green Version]
- Giordano, D.; Draves, K.E.; Young, L.B.; Roe, K.; Bryan, M.A.; Dresch, C.; Richner, J.M.; Diamond, M.S.; Gale, M.; Clark, E.A. Protection of mice deficient in mature B cells from West Nile virus infection by passive and active immunization. PLoS Pathog. 2017, 13, e1006743. [Google Scholar] [CrossRef]
- Mehlhop, E.; Ansarah-Sobrinho, C.; Johnson, S.; Engle, M.; Fremont, D.H.; Pierson, T.C.; Diamond, M.S. Complement protein C1q inhibits antibody-dependent enhancement of flavivirus infection in an IgG subclass-specific manner. Cell Host Microbe 2007, 2, 417–426. [Google Scholar] [CrossRef]
- Roe, K.; Giordano, D.; Young, L.B.; Draves, K.E.; Holder, U.; Suthar, M.S.; Gale, M.; Clark, E.A. Dendritic cell-associated MAVS is required to control West Nile virus replication and ensuing humoral immune responses. PLoS ONE 2019, 14, e0218928. [Google Scholar] [CrossRef]
- Sitati, E.; McCandless, E.E.; Klein, R.S.; Diamond, M.S. CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J. Virol. 2007, 81, 9801–9811. [Google Scholar] [CrossRef]
- Sitati, E.M.; Diamond, M.S. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J. Virol. 2006, 80, 12060–12069. [Google Scholar] [CrossRef]
- Shrestha, B.; Diamond, M.S. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef]
- Shrestha, B.; Diamond, M.S. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J. Virol. 2007, 81, 11749–11757. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Pinto, A.K.; Green, S.; Bosch, I.; Diamond, M.S. CD8+ T cells use TRAIL to restrict West Nile virus pathogenesis by controlling infection in neurons. J. Virol. 2012, 86, 8937–8948. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Lin, E.; Zhang, B.; Luster, A.D.; Tollett, J.; Samuel, M.A.; Engle, M.; Diamond, M.S. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005, 79, 11457–11466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chan, Y.K.; Lu, B.; Diamond, M.S.; Klein, R.S. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J. Immunol. 2008, 180, 2641–2649. [Google Scholar] [CrossRef]
- McCandless, E.E.; Wang, Q.; Woerner, B.M.; Harper, J.M.; Klein, R.S. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J. Immunol. 2006, 177, 8053–8064. [Google Scholar] [CrossRef]
- McCandless, E.E.; Zhang, B.; Diamond, M.S.; Klein, R.S. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc. Natl. Acad. Sci. USA 2008, 105, 11270–11275. [Google Scholar] [CrossRef] [Green Version]
- Acharya, D.; Wang, P.; Paul, A.M.; Dai, J.; Gate, D.; Lowery, J.E.; Stokic, D.S.; Leis, A.A.; Flavell, R.A.; Town, T.; et al. Interleukin-17A Promotes CD8+ T Cell Cytotoxicity To Facilitate West Nile Virus Clearance. J. Virol. 2017, 91, e01529-16. [Google Scholar] [CrossRef]
- Wang, Y.; Lobigs, M.; Lee, E.; Mullbacher, A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J. Virol. 2003, 77, 13323–13334. [Google Scholar] [CrossRef]
- Szretter, K.J.; Daniels, B.P.; Cho, H.; Gainey, M.D.; Yokoyama, W.M.; Gale, M.; Virgin, H.W.; Klein, R.S.; Sen, G.C.; Diamond, M.S. 2′-O Methylation of the Viral mRNA Cap by West Nile Virus Evades Ifit1-Dependent and -Independent Mechanisms of Host Restriction In Vivo. PLoS Pathog. 2012, 8, e1002698. [Google Scholar] [CrossRef]
- Brien, J.D.; Uhrlaub, J.L.; Nikolich-Žugich, J. West Nile virus-specific CD4 T cells exhibit direct antiviral cytokine secretion and cytotoxicity and are sufficient for antiviral protection. J. Immunol. 2008, 181, 8568–8575. [Google Scholar] [CrossRef]
- Durrant, D.M.; Robinette, M.L.; Klein, R.S. IL-1R1 is required for dendritic cell–mediated T cell reactivation within the CNS during West Nile virus encephalitis. J. Exp. Med. 2013, 210, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Ng, T.; Chu, H.J.; Noll, M.; Diamond, M.S. The relative contribution of antibody and CD8+ T cells to vaccine immunity against West Nile encephalitis virus. Vaccine 2008, 26, 2020–2033. [Google Scholar] [CrossRef] [PubMed]
- Lanteri, M.C.; O’Brien, K.M.; Purtha, W.E.; Cameron, M.J.; Lund, J.M.; Owen, R.E.; Heitman, J.W.; Custer, B.; Hirschkorn, D.F.; Tobler, L.H.; et al. Tregs control the development of symptomatic West Nile virus infection in humans and mice. J. Clin. Investig. 2009, 119, 3266–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, J.B.; Da Costa, A.; Lund, J.M. Regulatory T cells shape the resident memory T cell response to virus infection in the tissues. J. Immunol. 2014, 192, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, A.; Garza, E.; Graham, J.B.; Swarts, J.L.; Soerens, A.G.; Gale, M.; Lund, J.M. Extrinsic MAVS signaling is critical for Treg maintenance of Foxp3 expression following acute flavivirus infection. Sci. Rep. 2017, 7, 40720. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Luo, H.; Pang, L.; Peng, B.-H.; Winkelmann, E.; McGruder, B.; Hesse, J.; Whiteman, M.; Campbell, G.; Milligan, G.N.; et al. Dysregulation of Toll-Like Receptor 7 Compromises Innate and Adaptive T Cell Responses and Host Resistance to an Attenuated West Nile Virus Infection in Old Mice. J. Virol. 2016, 90, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, B.; Gottlieb, D.; Diamond, M.S. Infection and Injury of Neurons by West Nile Encephalitis Virus. J. Virol. 2003, 77, 13203–13213. [Google Scholar] [CrossRef]
- Philpott, D.C.E.; Nolan, M.S.; Evert, N.; Mayes, B.; Hesalroad, D.; Fonken, E.; Murray, K.O. Acute and Delayed Deaths after West Nile Virus Infection, Texas, USA, 2002–2012. Emerg. Infect. Dis. 2019, 25. [Google Scholar] [CrossRef]
- Diamond, M.S.; Klein, R.S. West Nile virus: Crossing the blood-brain barrier. Nat. Med. 2004, 10, 1294–1295. [Google Scholar] [CrossRef]
- Dai, J.; Wang, P.; Bai, F.; Town, T.; Fikrig, E. ICAM-1 participates in the entry of West Nile virus into the central nervous system. J. Virol. 2008, 82, 4164–4168. [Google Scholar] [CrossRef]
- Wang, P.; Dai, J.; Bai, F.; Kong, K.-F.; Wong, S.J.; Montgomery, R.R.; Madri, J.A.; Fikrig, E. Matrix Metalloproteinase 9 Facilitates West Nile Virus Entry into the Brain. J. Virol. 2008, 82, 8978–8985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arjona, A.; Foellmer, H.G.; Town, T.; Leng, L.; McDonald, C.; Wang, T.; Wong, S.J.; Montgomery, R.R.; Fikrig, E.; Bucala, R. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J. Clin. Investig. 2007, 117, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Brooke, C.B.; Whitmore, A.C.; Johnston, R.E. The Role of the Blood-Brain Barrier during Venezuelan Equine Encephalitis Virus Infection. J. Virol. 2011, 85, 10682–10690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.N.; Kent, K.A.; Bennett, C.J.; Bernard, K.A. Tissue tropism and neuroinvasion of West Nile virus do not differ for two mouse strains with different survival rates. Virology 2007, 368, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nir, Y.; Beemer, A.; Goldwasser, R.A. West Nile Virus infection in mice following exposure to a viral aerosol. Br. J. Exp. Pathol. 1965, 46, 443–449. [Google Scholar] [PubMed]
- Bai, F.; Town, T.; Pradhan, D.; Cox, J.; Ashish Ledizet, M.; Anderson, J.F.; Flavell, R.A.; Krueger, J.K.; Koski, R.A.; Fikrig, E. Antiviral peptides targeting the west nile virus envelope protein. J. Virol. 2007, 81, 2047–2055. [Google Scholar] [CrossRef]
- Suen, W.W.; Prow, N.A.; Hall, R.A.; Bielefeldt-Ohmann, H. Mechanism of West Nile Virus Neuroinvasion: A Critical Appraisal. Viruses 2014, 6, 2796–2825. [Google Scholar] [CrossRef]
- Cantile, C.; Del Piero, F.; Di Guardo, G.; Arispici, M. Pathologic and immunohistochemical findings in naturally occuring West Nile virus infection in horses. Vet. Pathol. 2001, 38, 414–431. [Google Scholar] [CrossRef]
- Guarner, J.; Shieh, W.-J.; Hunter, S.; Paddock, C.D.; Morken, T.; Campbell, G.L.; Marfin, A.A.; Zaki, S.R. Clinicopathologic study and laboratory diagnosis of 23 cases with West Nile virus encephalomyelitis. Hum. Pathol. 2004, 35, 983–990. [Google Scholar] [CrossRef]
- Wang, S.; Welte, T.; McGargill, M.; Town, T.; Thompson, J.; Anderson, J.F.; Flavell, R.A.; Fikrig, E.; Hedrick, S.M.; Wang, T. Drak2 contributes to West Nile virus entry into the brain and lethal encephalitis. J. Immunol. 2008, 181, 2084–2091. [Google Scholar] [CrossRef]
- Cardosa, M.J. Complement receptor mediates enhanced flavivirus replication in macrophages. J. Exp. Med. 1983, 158, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Diamond, M.S. Pathogenesis of West Nile Virus Infection: A Balance between Virulence, Innate and Adaptive Immunity, and Viral Evasion. J. Virol. 2006, 80, 9349–9360. [Google Scholar] [CrossRef]
- Brehin, A.C.; Mouries, J.; Frenkiel, M.P.; Dadaglio, G.; Despres, P.; Lafon, M.; Couderc, T. Dynamics of immune cell recruitment during West Nile encephalitis and identification of a new CD19+B220-BST-2+ leukocyte population. J. Immunol. 2008, 180, 6760–6767. [Google Scholar] [CrossRef]
- Rawal, A.; Gavin, P.J.; Sturgis, C.D. Cerebrospinal fluid cytology in seasonal epidemic West Nile virus meningo-encephalitis. Diagn. Cytopathol. 2006, 34, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L.; Pape, J.; Goody, R.J.; Corkill, M.; Kleinschmidt-DeMasters, B.K. CSF findings in 250 patients with serologically confirmed West Nile virus meningitis and encephalitis. Neurology 2006, 66, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-S.; Ramanathan, M.P.; Muthumani, K.; Choo, A.Y.; Jin, S.-H.; Yu, Q.-C.; Hwang, D.S.; Choo, D.K.; Lee, M.D.; Dang, K.; et al. Induction of Inflammation by West Nile virus Capsid through the Caspase-9 Apoptotic Pathway. Emerg. Infect. Dis. 2002, 8, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Parquet, M.D.C.; Kumatori, A.; Hasebe, F.; Morita, K.; Igarashi, A. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 2001, 500, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Cheeran, M.C.-J.; Hu, S.; Sheng, W.S.; Rashid, A.; Peterson, P.K.; Lokensgard, J.R. Differential responses of human brain cells to West Nile virus infection. J. Neurovirol. 2005, 11, 512–524. [Google Scholar] [CrossRef]
- Quick, E.D.; Leser, J.S.; Clarke, P.; Tyler, K.L. Activation of Intrinsic Immune Responses and Microglial Phagocytosis in an Ex Vivo Spinal Cord Slice Culture Model of West Nile Virus Infection. J. Virol. 2014, 88, 13005–13014. [Google Scholar] [CrossRef]
- Glass, W.G.; Lim, J.K.; Cholera, R.; Pletnev, A.G.; Gao, J.-L.; Murphy, P.M. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 2005, 202, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- Vig, P.J.S.; Lu, D.; Paul, A.M.; Kuwar, R.; Lopez, M.; Stokic, D.S.; Leis, A.A.; Garrett, M.R.; Bai, F. Differential Expression of Genes Related to Innate Immune Responses in Ex Vivo Spinal Cord and Cerebellar Slice Cultures Infected with West Nile Virus. Brain Sci. 2018, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Armah, H.B.; Wang, G.; Omalu, B.I.; Tesh, R.B.; Gyure, K.A.; Chute, D.J.; Smith, R.D.; Dulai, P.; Vinters, H.V.; Kleinschmidt-DeMasters, B.K.; et al. Systemic Distribution of West Nile Virus Infection: Postmortem Immunohistochemical Study of Six Cases. Brain Pathol. 2007, 17, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Leis, A.A.; Stokic, D.S.; Petzold, A. Glial S100B is elevated in serum across the spectrum of west nile virus infection. Muscle Nerve 2012, 45, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Groves, M.; Leis, A.; Scaravilli, F.; Stokic, D.; Stokic, D. Neuronal and glial cerebrospinal fluid protein biomarkers are elevated after West Nile Virus infection. Muscle Nerve 2010, 41, 42–49. [Google Scholar] [CrossRef]
- Kuwar, R.; Stokic, D.; Leis, A.; Bai, F.; Paul, A.; Fratkin, J.; Vig, P.; Paul, A. Does astroglial protein S100B contribute to West Nile neuro-invasive syndrome? J. Neurol. Sci. 2015, 358, 243–252. [Google Scholar] [CrossRef]
- Chu, J.J.H.; Ng, M.L. The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J. Gen. Virol. 2003, 84, 3305–3314. [Google Scholar] [CrossRef]
- Wang, H.; Ward, M.F.; Fan, X.-G.; Sama, A.E.; Li, W. Potential role of high mobility group box 1 in viral infectious diseases. Viral Immunol. 2006, 19, 3–9. [Google Scholar] [CrossRef]
- Juranek, J.; Ray, R.; Banach, M.; Rai, V. Receptor for advanced glycation end-products in neurodegenerative diseases. Rev. Neurosci. 2015, 26, 691–698. [Google Scholar] [CrossRef]
- Kumar, M.; Verma, S.; Nerurkar, V.R. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflamm. 2010, 7, 73. [Google Scholar] [CrossRef]
- Verma, S.; Kumar, M.; Nerurkar, V.R. Cyclooxygenase-2 inhibitor blocks the production of West Nile virus-induced neuroinflammatory markers in astrocytes. J. Gen. Virol. 2011, 92, 507–515. [Google Scholar] [CrossRef]
- Murray, K.O.; Garcia, M.N.; Rahbar, M.H.; Martínez, D.; Khuwaja, S.A.; Arafat, R.R.; Rossmann, S. Survival Analysis, Long-Term Outcomes, and Percentage of Recovery up to 8 Years Post-Infection among the Houston West Nile Virus Cohort. PLoS ONE 2014, 9, e102953. [Google Scholar] [CrossRef] [PubMed]
- Weatherhead, J.E.; Miller, V.E.; Garcia, M.N.; Hasbun, R.; Salazar, L.; Dimachkie, M.M.; Murray, K.O. Long-term neurological outcomes in West Nile virus-infected patients: An observational study. Am. J. Trop. Med. Hyg. 2015, 92, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.O.; Nolan, M.S.; Ronca, S.E.; Datta, S.; Govindarajan, K.; Narayana, P.A.; Salazar, L.; Woods, S.P.; Hasbun, R. The Neurocognitive and MRI Outcomes of West Nile Virus Infection: Preliminary Analysis Using an External Control Group. Front. Neurol. 2018, 9, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leis, A.A.; Szatmary, G.; Ross, M.A.; Stokic, D.S. West nile virus infection and myasthenia gravis. Muscle Nerve 2013, 49, 26–29. [Google Scholar] [CrossRef]
- Hawkes, M.A.; Hocker, S.E.; Leis, A.A. West Nile virus induces a post-infectious pro-inflammatory state that explains transformation of stable ocular myasthenia gravis to myasthenic crises. J. Neurol. Sci. 2018, 395, 1–3. [Google Scholar] [CrossRef]
- Mu, L.; Zhang, Y.; Sun, B.; Wang, J.; Xie, X.; Li, N.; Zhang, J.; Kong, Q.; Liu, Y.; Han, Z.; et al. Activation of the receptor for advanced glycation end products (RAGE) exacerbates experimental autoimmune myasthenia gravis symptoms. Clin. Immunol. 2011, 141, 36–48. [Google Scholar] [CrossRef]
- Moser, B.; Bekos, C.; Zimprich, F.; Nickl, S.; Klepetko, W.; Ankersmit, J. The receptor for advanced glycation endproducts and its ligands in patients with myasthenia gravis. Biochem. Biophys. Res. Commun. 2012, 420, 96–101. [Google Scholar] [CrossRef]
- Xie, Y.; Li, H.-F.; Jiang, B.; Li, Y.; Kaminski, H.J.; Kusner, L.L. Elevated plasma interleukin-17A in a subgroup of Myasthenia Gravis patients. Cytokine 2016, 78, 44–46. [Google Scholar] [CrossRef]
- Schaffert, H.; Pelz, A.; Saxena, A.; Losen, M.; Meisel, A.; Thiel, A.; Kohler, S. IL-17-producing CD4(+) T cells contribute to the loss of B-cell tolerance in experimental autoimmune myasthenia gravis. Eur. J. Immunol. 2015, 45, 1339–1347. [Google Scholar] [CrossRef]
- Ahmed, S.; Libman, R.; Wesson, K.; Ahmed, F.; Einberg, K. Guillain-Barre syndrome: An unusual presentation of West Nile virus infection. Neurology 2000, 55, 144–146. [Google Scholar] [CrossRef]
- Almhanna, K.; Palanichamy, N.; Sharma, M.; Hobbs, R.; Sil, A. Unilateral Brachial Plexopathy Associated with West Nile Virus Meningoencephalitis. Clin. Infect. Dis. 2003, 36, 1629–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassin-Baer, S.; Kirson, E.D.; Shulman, L.; Buchman, A.S.; Bin, H.; Hindiyeh, M.; Markevich, L.; Mendelson, E. Stiff-Person Syndrome Following West Nile Fever. Arch. Neurol. 2004, 61, 938–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagianni, P.; Alexopoulos, H.; Sourdi, A.; Papadimitriou, D.; Dimitrakopoulos, A.; Moutsopoulos, H. West Nile Virus infection triggering autoimmune encephalitis: Pathophysiological and therapeutic implications. Clin. Immunol. 2019, 207, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Ulbert, S. West Nile virus vaccines-current situation and future directions. Hum. Vaccines Immunother. 2019, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswami, P.E.L.; Hyde, C.; Page, C.; Hastings, N.E. West Nile meningitis/ encephalitis: Experience with corticosteroid therapy. Neurology 2004, 62, A404. [Google Scholar]
- Pyrgos, V.; Younus, F. High-dose steroids in the management of acute flaccid paralysis due to West Nile virus infection. Scand. J. Infect. Dis. 2004, 36, 509–512. [Google Scholar] [CrossRef]
- Leis, A.A.; Sinclair, D.J. Lazarus Effect of High Dose Corticosteroids in a Patient with West Nile Virus Encephalitis: A Coincidence or a Clue? Front. Med. 2019, 6, 81. [Google Scholar] [CrossRef]
- Huang, C.; Slater, B.; Rudd, R.; Parchuri, N.; Hull, R.; Dupuis, M.; Hindenburg, A. First Isolation of West Nile virus from a Patient with Encephalitis in the United States. Emerg. Infect. Dis. 2002, 8, 1367–1371. [Google Scholar] [CrossRef]
- Bandara, S.R.; Herath, H. Effectiveness of corticosteroid in the treatment of dengue—A systemic review. Heliyon 2018, 4, e00816. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, F.; Thompson, E.A.; Vig, P.J.S.; Leis, A.A. Current Understanding of West Nile Virus Clinical Manifestations, Immune Responses, Neuroinvasion, and Immunotherapeutic Implications. Pathogens 2019, 8, 193. https://doi.org/10.3390/pathogens8040193
Bai F, Thompson EA, Vig PJS, Leis AA. Current Understanding of West Nile Virus Clinical Manifestations, Immune Responses, Neuroinvasion, and Immunotherapeutic Implications. Pathogens. 2019; 8(4):193. https://doi.org/10.3390/pathogens8040193
Chicago/Turabian StyleBai, Fengwei, E. Ashley Thompson, Parminder J. S. Vig, and A. Arturo Leis. 2019. "Current Understanding of West Nile Virus Clinical Manifestations, Immune Responses, Neuroinvasion, and Immunotherapeutic Implications" Pathogens 8, no. 4: 193. https://doi.org/10.3390/pathogens8040193